





Docking ligand libraries can be a daunting task for molecular modelers, especially when dealing with large datasets and complex configurations. Fortunately, the AutoDock Vina Extended Extension in SAMSON provides an efficient and user-friendly way to tackle this challenge. This post explores how to streamline the ligand library docking process, leveraging key features of this extension to reduce effort and enhance results.
The Challenge: Managing Large Ligand Libraries
When working with a dataset of compounds, generating meaningful receptor-ligand binding poses requires accurate and reproducible methods. A common pain point is ensuring that ligands are configured correctly and search domains are well-defined. Manual setups can lead to inconsistencies and runtime inefficiencies. This is where tools for automating complex workflows, such as SAMSON’s AutoDock Vina Extended, become invaluable.
The Solution: AutoDock Vina Extended
The AutoDock Vina Extended Extension in SAMSON bridges the gap by offering a simple yet powerful interface to set up, execute, and analyze docking projects. Here’s a breakdown of how you can set up a ligand library docking process:
1. Configure Your Ligand Library
To start, select the Ligand library option in the Set ligand panel and browse to the directory containing your ligand files. AutoDock Vina Extended supports formats like MOL2 and SDF, with the ability to process multiple ligands per file.
If your ligands contain 2D structures or are missing hydrogens, it’s essential to minimize and preprocess them before docking. Check the Minimize option and ensure Add missing hydrogens is enabled. This ensures ligands are optimized for docking and reduces potential errors during computation.
2. Customize Rotatable Bonds
Flexibility in molecular structures plays an important role in accurate docking simulations. AutoDock Vina Extended allows you to control rotatable bonds for ligands. For instance, you can lock specific bond types by adjusting the Locked bonds settings and enabling the Lock specific ligand bonds option. This feature can substantially reduce docking time while preserving realistic molecular conformations.

3. Define the Search Domain
By default, the search domain is based on the receptor you select. However, you can refine this area to faster target binding sites and improve efficiency. Use options such as Based on pocket/selection or manually adjust the domain box in the Viewport using its corner controllers. For a ligand library docking, defining a smaller search domain surrounding the known active site can greatly speed up computations.

4. Run the Docking Process
Once your preparation steps are complete, start docking by pressing the Dock library button. Customize advanced options such as Exhaustiveness and Maximum number of generated binding modes for tailored results. If needed, enable auto-saving to generate a comprehensive project directory containing results, ensuring reproducibility and easy access for future analyses.
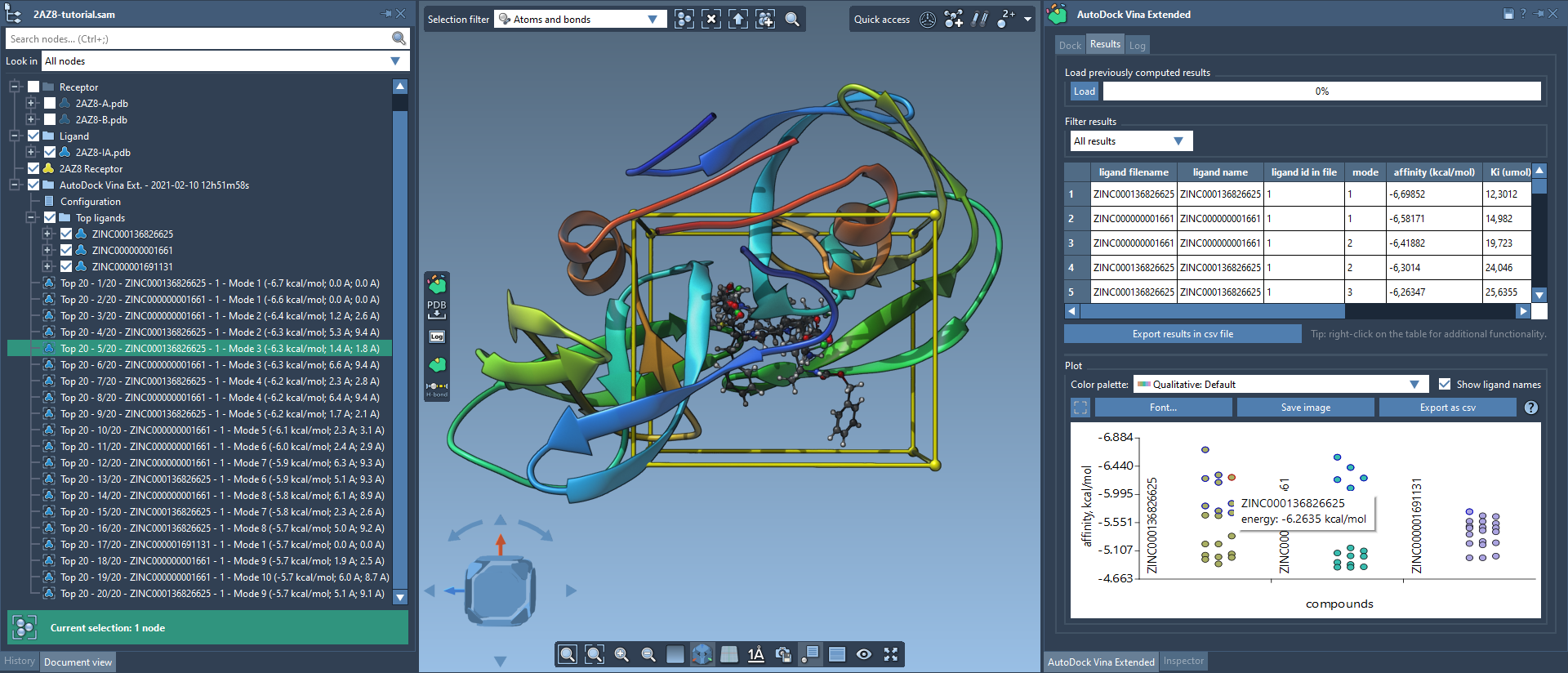
Streamlining Result Analysis
Post-docking, SAMSON offers robust tools for analyzing output data. The generated results folder organizes information into Top ligands and associated conformations. Additionally, the results table and interactive plots provide features such as filtering by affinity scores and exporting relevant data in CSV format for external analysis.
Conclusion
The AutoDock Vina Extended Extension in SAMSON simplifies ligand library docking with its seamless workflow and extensive customization for ligands, receptors, and search domains. By automating and optimizing the docking process, it empowers molecular modelers to focus on experimental insights.
For a detailed step-by-step guide, refer to the official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get started with SAMSON today by visiting SAMSON Connect.