





If you’ve ever struggled with modeling molecular systems that require frequent editing and topological changes, the Interactive Modeling Universal Force Field (IM-UFF) in SAMSON might be just what you need. Unlike traditional methods, IM-UFF is designed to handle interactive editing with ease, allowing changes in topology, bond orders, and atom types—all while keeping the process smooth and intuitive.
Addressing the Modeling Challenge
In typical molecular modeling workflows, making changes to the system, such as forming or breaking bonds, can be cumbersome. Traditional force fields often aren’t equipped to handle such dynamic edits efficiently, leading to errors or requiring significant manual adjustments. IM-UFF extends the widely-used Universal Force Field (UFF) to simplify these tasks during interactive editing, helping molecular modelers focus on designing and refining structures rather than fixing modeling issues.
What Makes IM-UFF Unique?
IM-UFF introduces a seamless way to work with molecular topologies. Here’s what sets it apart:
- Dynamic Topology Updates: IM-UFF smoothly adapts to topological changes. Whether you’re creating or breaking covalent bonds, adjusting bond orders, or modifying atom types, the interaction model intuitively updates these changes while preserving physical realism.
- Interactive Editing: It puts control directly in your hands, letting you move atoms and observe how bonds form or disconnect in real time.
- Configurability: IM-UFF offers customizable options for static and dynamic simulations, enabling workflows tailored to your project’s needs.

How to Set Up IM-UFF
Getting started with IM-UFF in SAMSON is straightforward:
- Open a molecular system in SAMSON.
- Add a simulator via
Edit > Simulate > Add simulatoror use the shortcutCtrl + Shift + M(on Windows) orCmd + Shift + M(on macOS). - Select Interactive Modeling Universal Force Field from the list of interaction models.
- Pick a state updater, such as the FIRE Engine, for optimizing geometry during the simulation.
- Start the simulation and adjust the parameters as needed in the IM-UFF parameter window.
The parameter window allows you to toggle options like static topology or exclude van der Waals forces for manipulated atoms, streamlining the editing experience.
Interactive Modeling in Action
With IM-UFF, real-time interactions become intuitive. For example, when an atom is displaced slightly, the system locally adjusts while preserving bonds. However, significant displacements result in bond breakage, and new topologies form dynamically when atoms approach others.
This creates a powerful environment for testing and evolving molecular systems, especially when compared to static force fields like standard UFF. You can experiment with small or medium systems first to observe how the topology behaves during manipulation—perfect for getting acquainted with the tool’s capabilities.
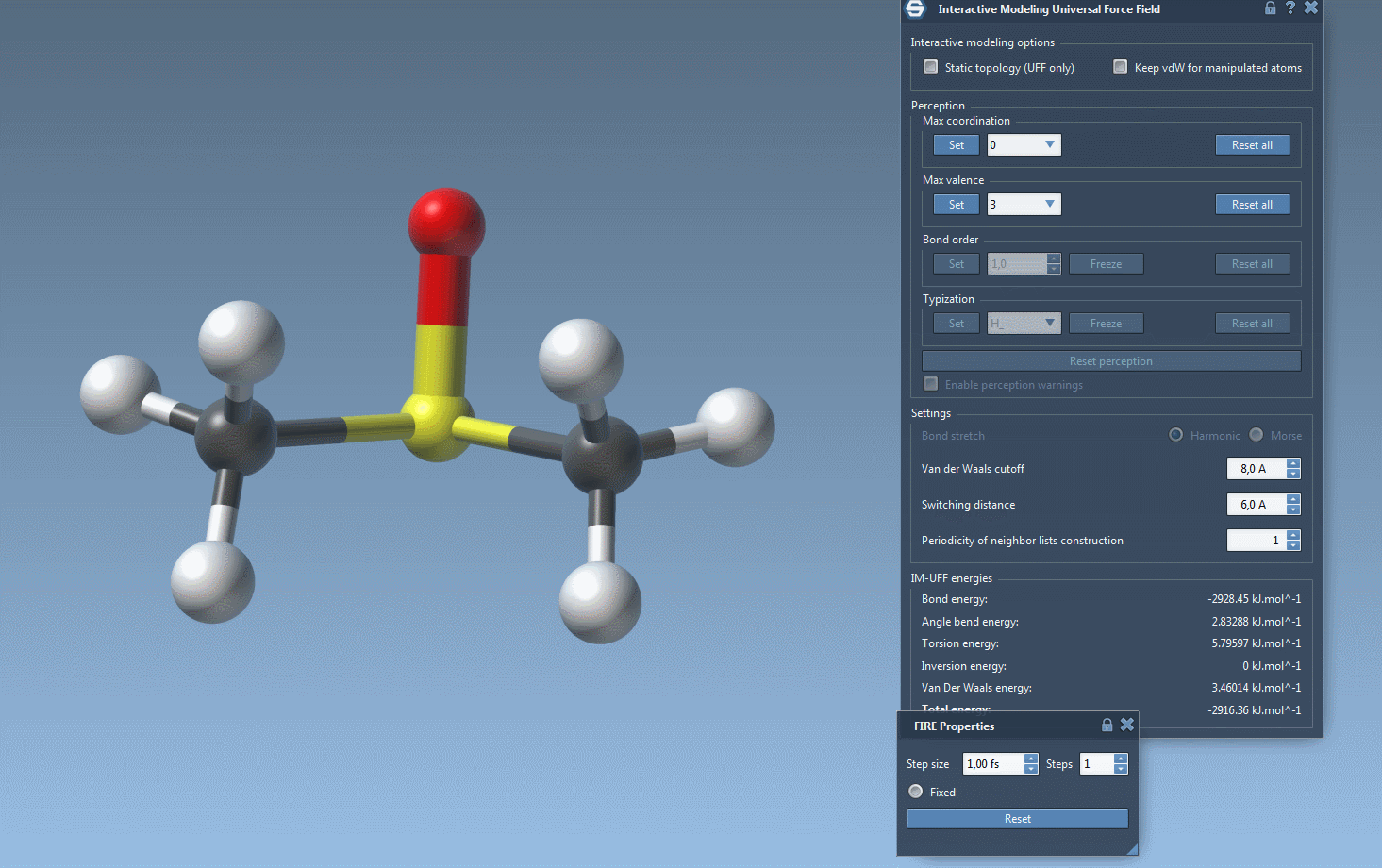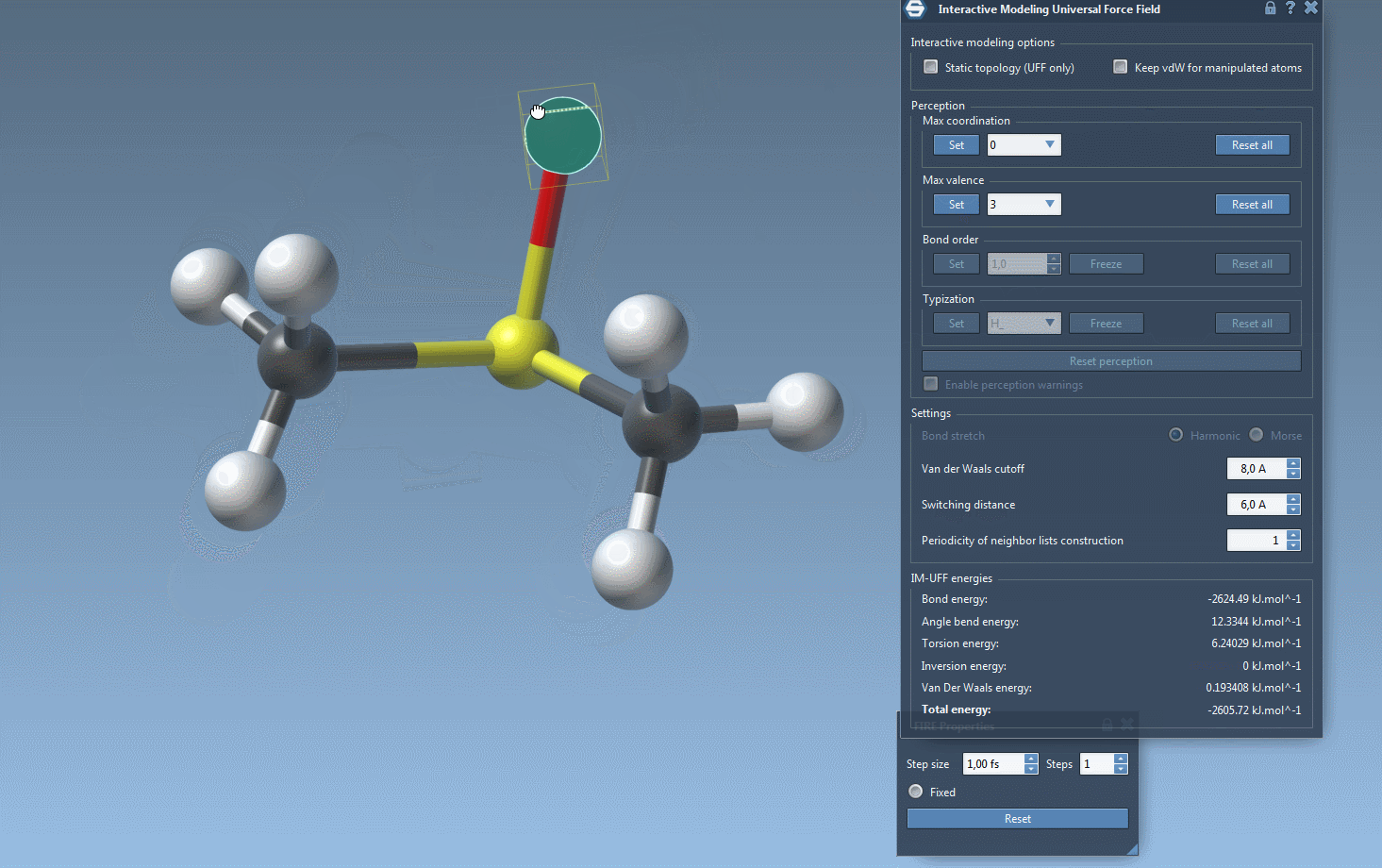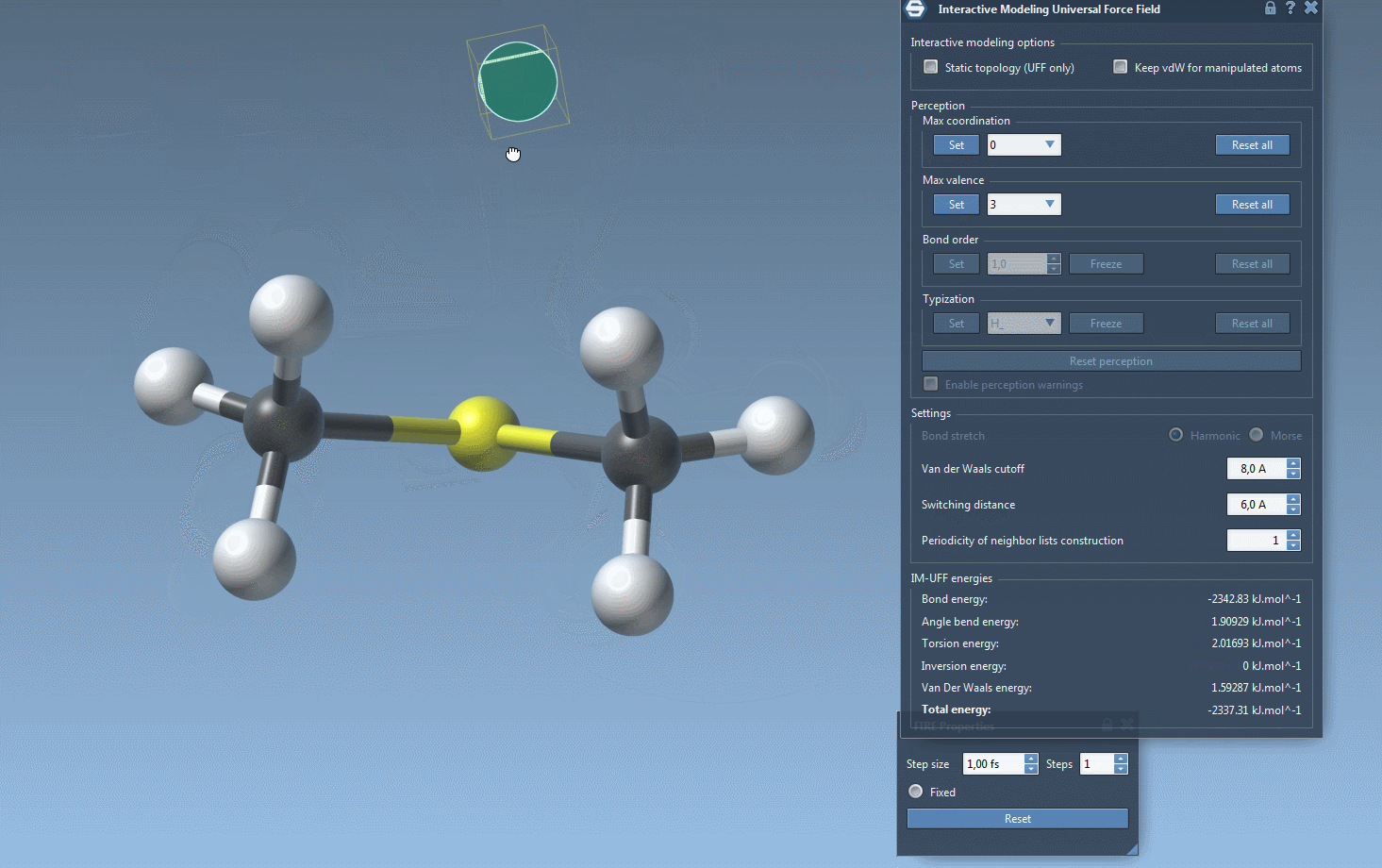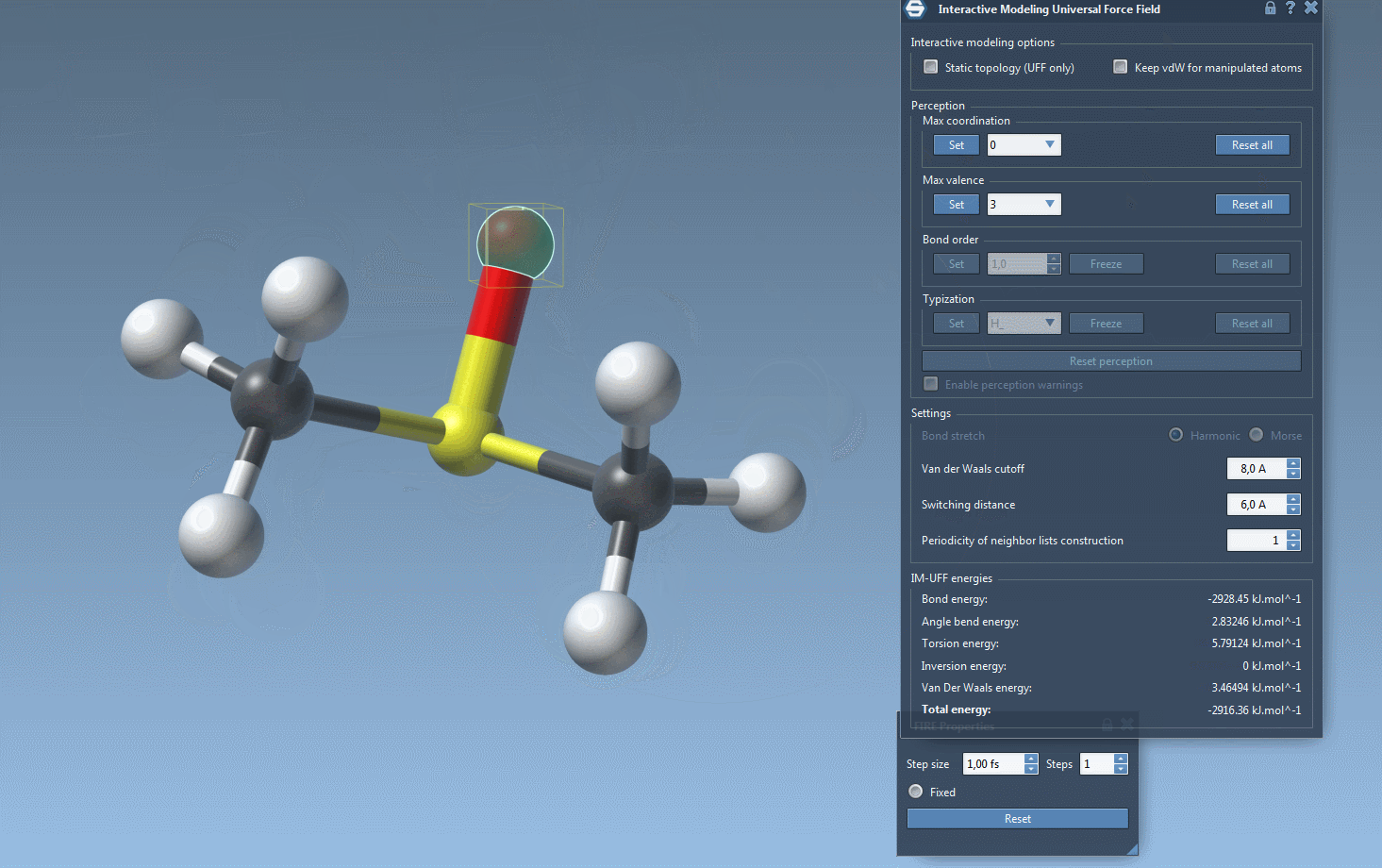
Customizing IM-UFF
IM-UFF is not a one-size-fits-all solution—it offers flexibility for different modeling needs. For example:
- Van der Waals Cutoff: Adjust this to refine how non-bonded interactions are computed.
- Maximum Coordination and Valence: Use these to control atom behavior during simulations.
Additionally, atoms can be added or removed dynamically, and their typizations are updated automatically. This ensures consistent results across all edits without manual recalibration.
Ready to Explore?
If you’re looking to simplify your molecular editing workflows or need a force field capable of dynamic adjustments, IM-UFF is worth exploring. Try it on a smaller molecular structure and compare its functionality against standard UFF to see the difference.
To learn more, visit the detailed IM-UFF documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON today at samson-connect.net.