





One of the challenges molecular modelers often face is accurately representing ligand flexibility during docking simulations. Flexible ligands are essential for realistic docking results, but managing rotatable bonds can dramatically increase computation times. In this blog post, we’ll delve into how to efficiently set up rotatable and locked bonds for ligands in SAMSON using the AutoDock Vina Extended Extension, ensuring accurate and time-effective simulations.
Why Ligand Flexibility Matters
During protein-ligand docking, the ability of ligands to explore different conformations significantly impacts binding affinity predictions. Adjusting ligand flexibility—through specifying which bonds can rotate and which remain fixed—brings a balance between computational efficiency and the realism of simulation results.
Step 1: Setting Up a Single Ligand
To get started, check the Single ligand option in the Set ligand section of the AutoDock Vina Extended interface. Select your desired ligand (e.g., “2AZ8-IA”) from the Document view and click on the Set button.
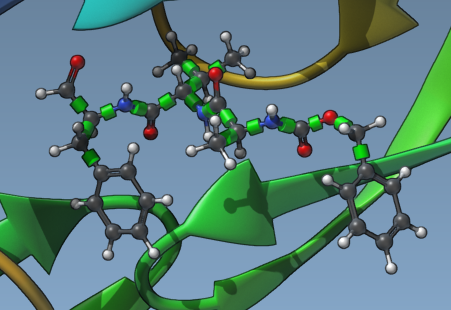
Step 2: Understanding Rotatable Bonds
Once your ligand is set, green cylinders will appear in the Viewport, overlaying rotatable bonds in your ligand model. These represent bond controllers for flexibility. By clicking on these cylinders, you can toggle the status of each bond between rotatable (green) and non-rotatable (red). Managing rotatable bonds lets you control the ligand’s complexity and computation demand.
Step 3: Locking Specific Bonds
To streamline the process further, you can set Locked bonds, ensuring specific bond types remain non-rotatable throughout the docking simulation:
- In the AutoDock Vina Extended interface, click on Locked bonds settings.
- Check the bond types you wish to lock (e.g., double bonds or amide bonds).
- Check the Lock specific ligand bonds option under the Ligand Settings section.
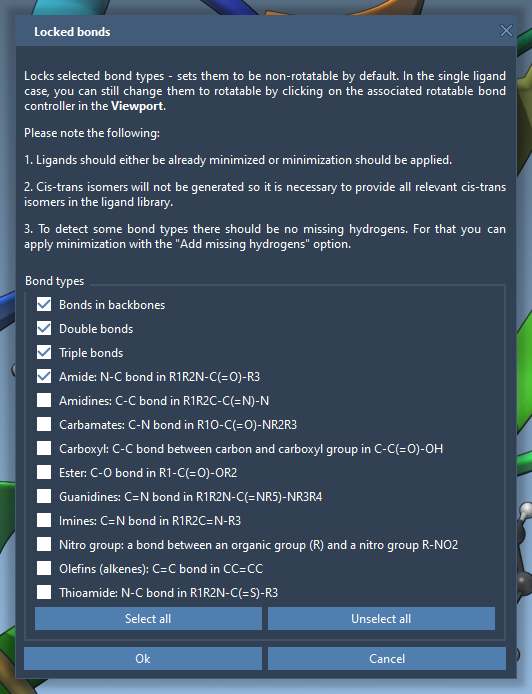
When locked bonds are set, relevant bond controllers will turn red to clearly indicate their non-rotatable state. This step allows you to avoid unnecessary sampling, reducing computational complexity.
Step 4: Minimization and Adding Hydrogens
If your ligands need preparation (e.g., are in 2D or missing hydrogens), you can use AutoDock Vina Extended’s minimization tools:
- Check the Minimize option before docking.
- Enable Add missing hydrogens to ensure polar hydrogens are accounted for, critical for hydrogen bond detection and docking accuracy.
The minimization stops automatically based on energy differences or stepping thresholds, depending on your settings. For high-confidence results, ensure ligands are properly minimized.

Optimized Docking, Realistic Results
By strategically managing ligand flexibility through rotatable bonds, locked bonds, and minimization, you can confidently achieve realistic and efficient protein-ligand docking simulations. With these insights, tackling complex docking scenarios becomes more straightforward and computationally feasible.
Want to dive deeper into ligand flexibility setup and other tools offered by AutoDock Vina Extended? Check out the full documentation at this link.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON at https://www.samson-connect.net.