





Molecular modelers often face the challenge of setting up simulations quickly while ensuring accuracy. If you’ve worked with force fields, you know that a precise setup is crucial for achieving meaningful results. SAMSON, the integrative molecular design platform, takes this process a step further with the Universal Force Field (UFF). Whether you’re new to SAMSON or an experienced modeler, this post walks you through simplifying your simulation workflows with UFF.
Why Use UFF?
The Universal Force Field (UFF), proposed by Rappe et al., is a comprehensive interaction model that spans the periodic table. Its implementation in SAMSON goes beyond basic force field setups by integrating automatic molecule perception. This means bonds, bond orders, and atom types are computed automatically, enabling a much faster transition from molecular structure to simulation-ready system.
Setting Up UFF in SAMSON
Getting started with UFF is straightforward. Follow these steps to set up your simulation:
- Open a document containing the molecular system you wish to simulate.
- Add a simulator by navigating to
Edit > Simulate > Add simulator. Alternatively, use the shortcut:Ctrl + Shift + M(Windows) orCmd + Shift + M(Mac). - From the list of interaction models, select Universal Force Field.
- Choose a state updater for your simulation, such as the FIRE (Fast Inertial Relaxation Engine).
- Press OK to finalize the setup.
A dedicated UFF setup window will then appear:
- Select Use existing bonds if you want UFF to rely on predefined bonds from your molecule’s structure. If not checked, UFF will recompute bonds based on atomic types and positions.
- Click OK to proceed.
At this stage, SAMSON performs several preprocessing operations, which include:
- Calculating covalent bonds when Use existing bonds is not selected,
- Determining bond orders, and
- Assigning appropriate atom types.
If any inconsistencies are detected during this phase, a warning message will specify the problem. This ensures you can troubleshoot accurately and proceed confidently.
Interactivity at Your Fingertips
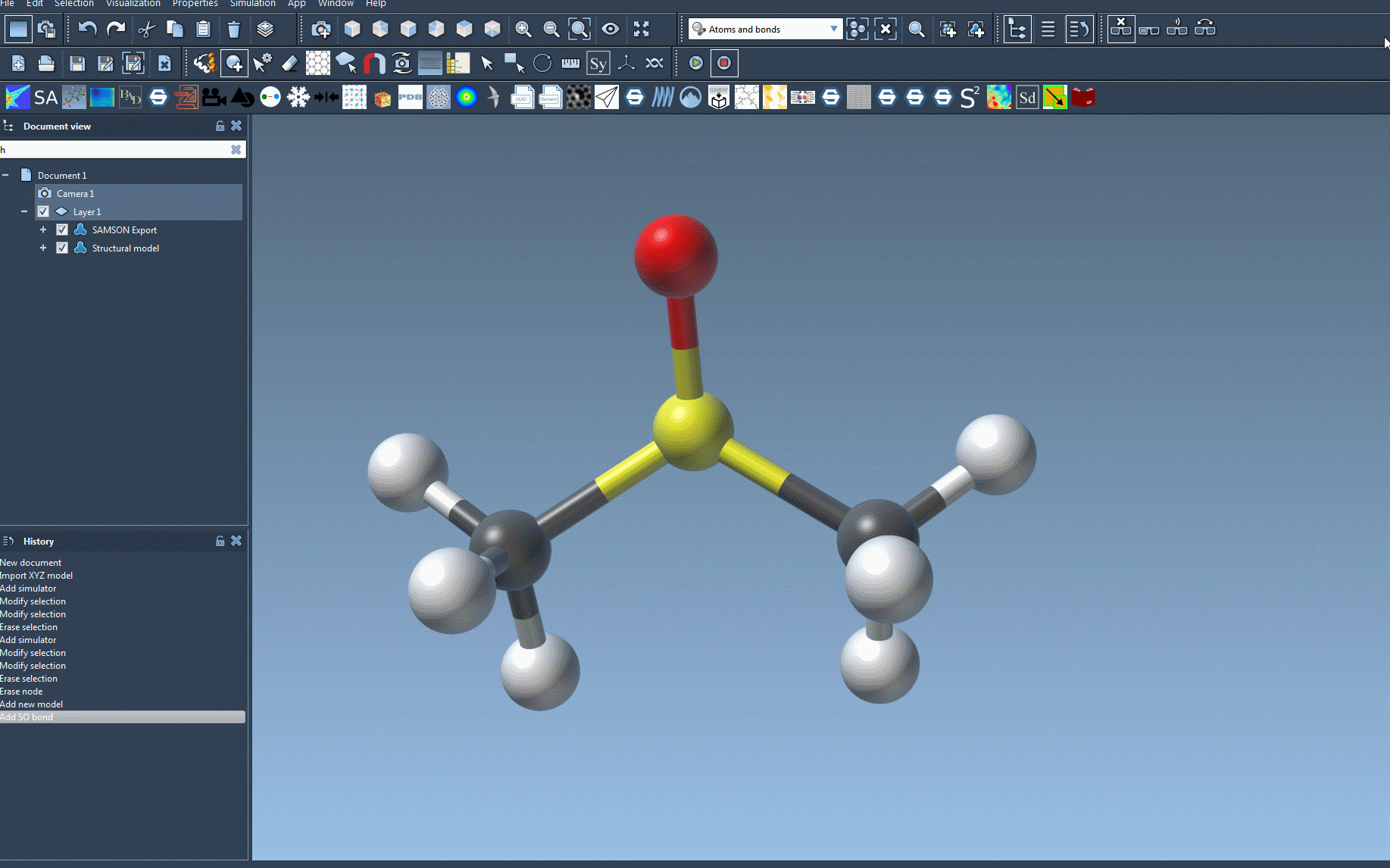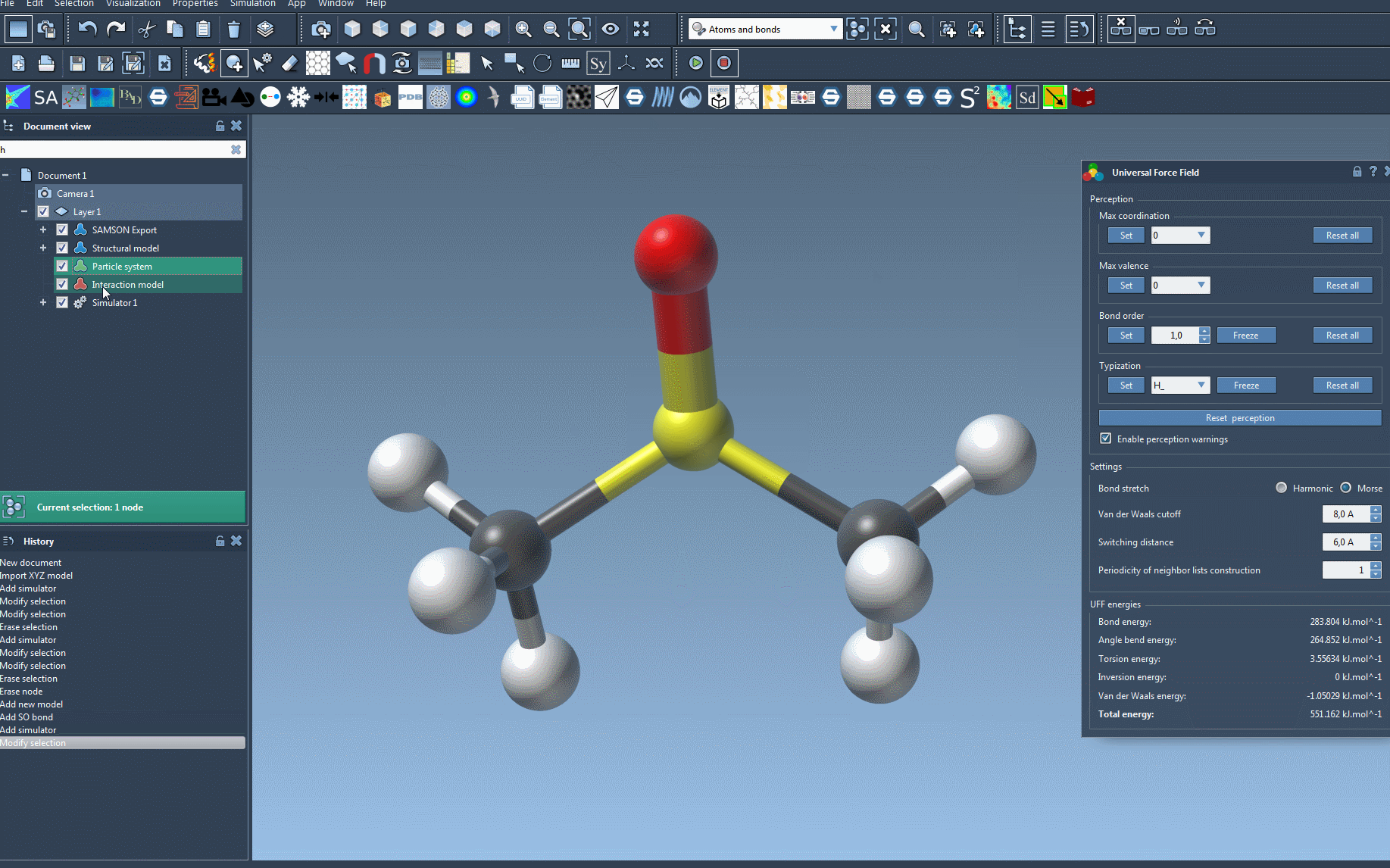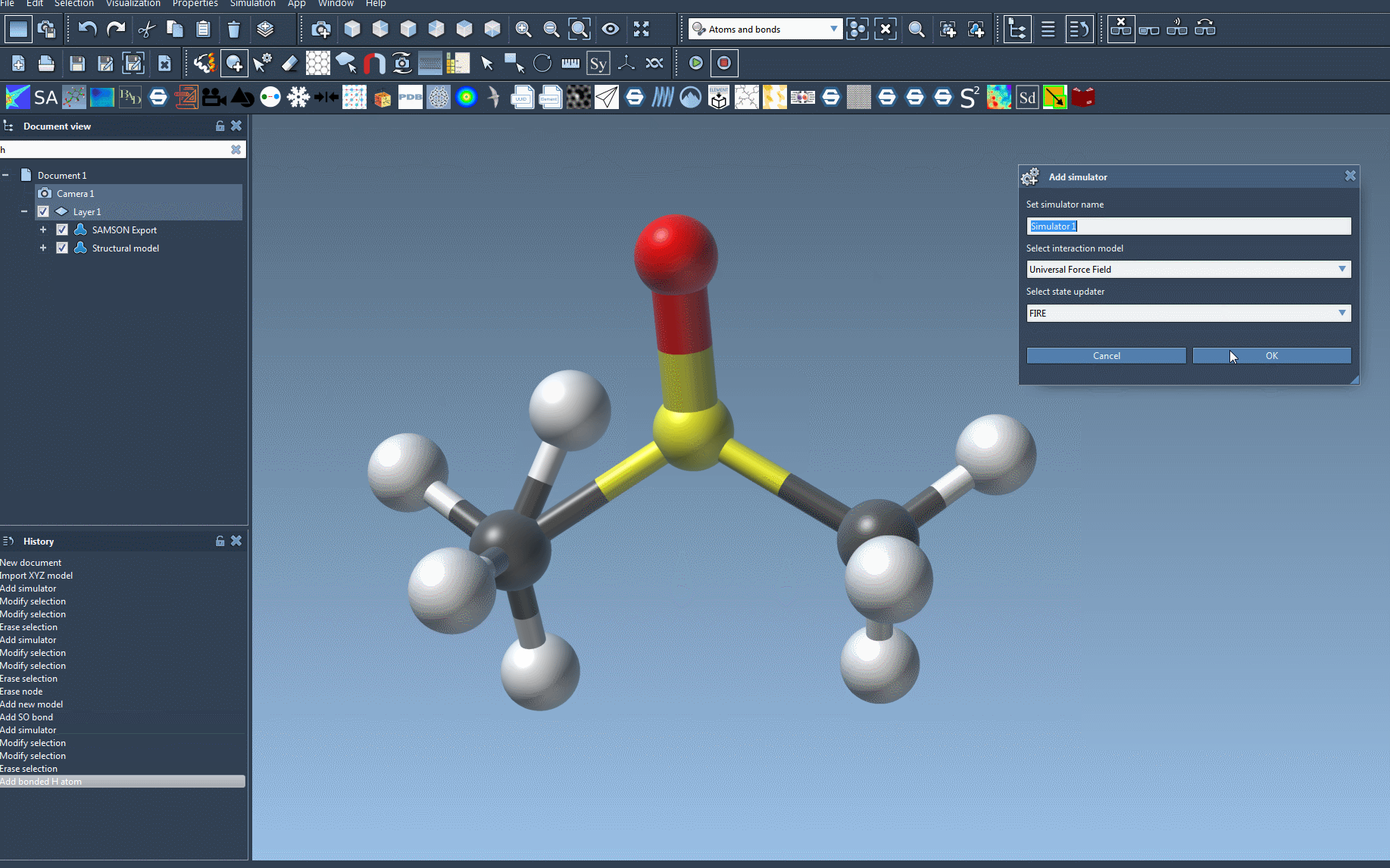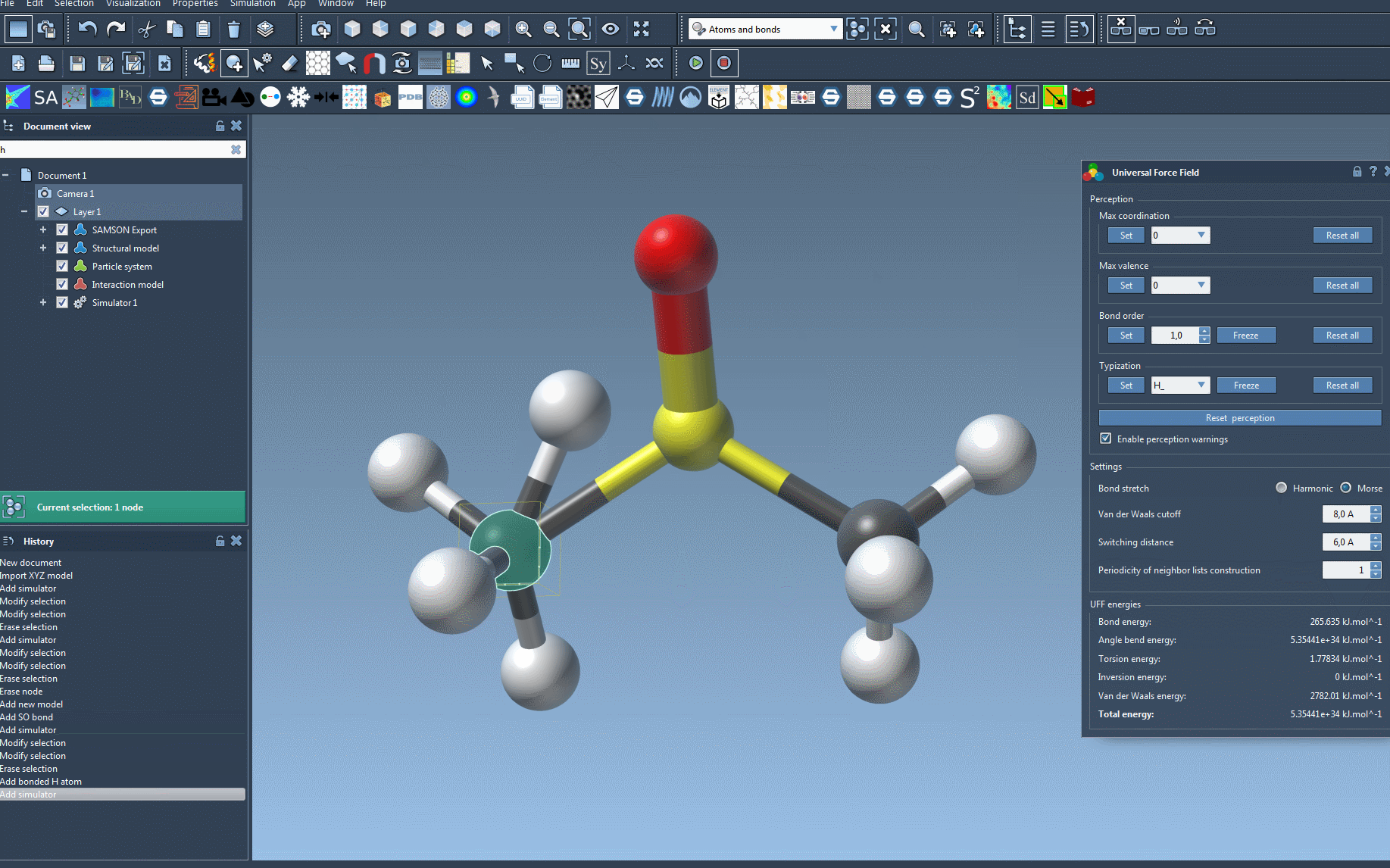
Once the setup is complete, head to the parameter window of UFF. Here, you can begin your simulation by selecting Edit > Simulate > Start. The interactive interface is one of UFF’s most powerful features—it allows you to:
- Modify the position of atoms in real time to observe the adjustments in neighboring atom positions,
- Analyze the UFF energy terms, displayed at the bottom of the window, including the total energy and individual contributions,
- Adjust key parameters like bond-stretch interactions (e.g., switch between Harmonic and Morse potential), van der Waals cutoff distances, and periodicity for neighbor list construction.
Carefully tailoring these parameters can help you achieve optimized results for specific molecular systems.
Conclusion
UFF’s streamlined setup in SAMSON removes much of the complexity traditionally associated with force field simulations. Ready to get started? Dive deeper into the setup and its customization options by visiting the official documentation at this page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at SAMSON Connect.