





Docking studies are vital for molecular modeling, especially when exploring protein-ligand interactions. One persistent challenge is accounting for molecular flexibility—many docking protocols assume rigid receptor structures, which can overlook biologically relevant conformations. With AutoDock Vina Extended as part of the SAMSON platform, you can now incorporate flexible receptor side chains seamlessly. Here’s how to get started and why it matters for improving docking precision.
Why Flexible Side Chains Matter
Protein structures aren’t static. In biological systems, side chains within the binding pocket can adapt to accommodate ligands. Ignoring this flexibility may result in unrealistic docking poses or missed potential hits. Incorporating adjustable side chains can yield better biologically meaningful results in important cases where induced fit effects are significant.
The AutoDock Vina Extended SAMSON Extension makes it simple to define receptor flexibility during docking. This significantly enhances the accuracy of docking, albeit with increased computational complexity.
Step-by-Step: Setting Receptor Flexibility
Here’s a practical approach to incorporating flexible residues in your docking workflows:
1. Select Flexible Residues
You can identify and select residues around the binding site using different methods:
- In the Document View: Highlight residues from the receptor’s structural model and directly select them for flexibility.
- In the Viewport: Switch the Selection filter to Residues (in the top-left corner). Use Ctrl + Alt to add or remove individual residues.
- Select Based on the Binding Site: If the receptor has a bound ligand, select the ligand and navigate to Select > Biology > Binding sites. Define parameters like proximity and inclusion to specify the residues.
Each of these approaches provides flexibility depending on the complexity and nature of your receptor.
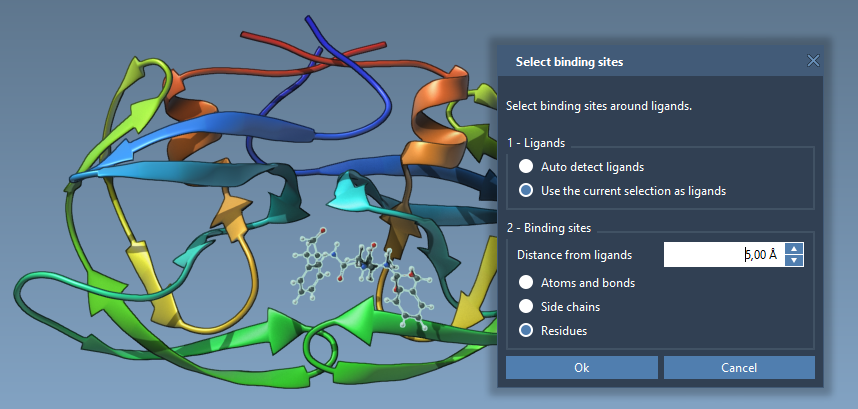
2. Set Receptor Flexibility
Once residues are selected, assign them as flexible:
- In the AutoDock Vina Extended, head to the Set receptor section.
- Click the Set button to apply your selection as flexible side chains.
In the Viewport, bonds of the flexible residues will be marked with green cylinders. These represent rotatable bonds. Clicking on a bond changes its state—green means rotatable, while red means locked. This granularity allows better control over flexibility without overloading computational resources.
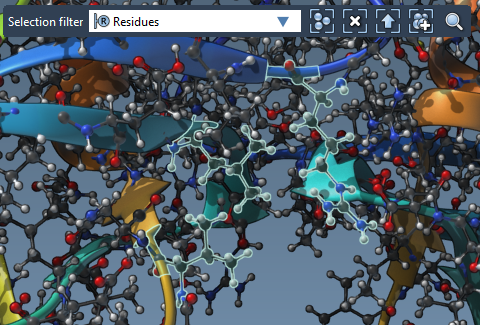
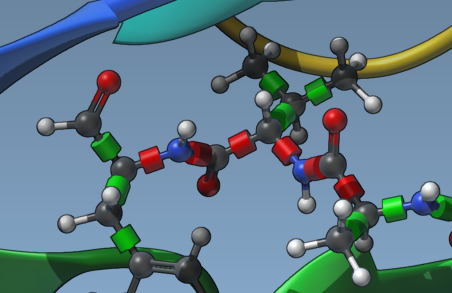
3. Refine Rotatable Bonds
Customizing flexibility doesn’t end with side-chain selection. You can fine-tune rotatable bonds:
- Click directly on bond controllers in the Viewport to toggle their flexibility state.
- Use the Locked bonds settings option to configure specific bond types (e.g., pi-bonds or double bonds) as permanently locked.
These features ensure computational precision while tailoring flexibility to the biological scenario you’re modeling.
Optimizing Results
Adding flexible chains demands additional computational resources and time. This trade-off ensures docking results reflect more biologically relevant receptor-ligand interactions. To minimize complexity:
- Limit the flexibility to residues immediately around the binding site.
- Leverage AutoDock Vina’s optimized algorithms for better performance-control balance.
Experiment with Confidence
Incorporating flexible receptor side chains elevates the quality of your docking studies. By accounting for receptor flexibility, especially around the binding pocket, you enhance prediction accuracy for ligand binding poses and energies. Try it today with the AutoDock Vina Extended in SAMSON to see the difference for yourself.
Learn more by exploring the full documentation at this link.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get started by downloading SAMSON at https://www.samson-connect.net.