





One of the major challenges in molecular docking is accurately accounting for ligand flexibility. Flexible ligands with rotatable bonds can enhance the reliability of docking results by adopting realistic conformations during the docking process. However, managing these flexible bonds can significantly increase computational demands if not handled efficiently. Thankfully, the AutoDock Vina Extended extension in SAMSON offers intuitive tools to set up, customize, and optimize ligand flexibility settings for docking experiments. Here’s how.
Setting Up Rotatable Bonds Step-by-Step
After selecting your ligand in SAMSON, rotatable bond controllers automatically appear in the Viewport. These controllers are represented as green cylinders superimposed over bonds, indicating they can rotate by default. You can visually inspect and manage these bonds to ensure a balance between computational efficiency and docking realism.
Zoom in on the ligand for a closer look by selecting it in the Document view and pressing Shift + Space. Clicking on the green cylinders toggles bonds between rotatable (green) and non-rotatable (red). This level of control lets you precisely tailor bond flexibility according to the specific needs of your docking experiment.
Automating Rotatable Bond Configurations
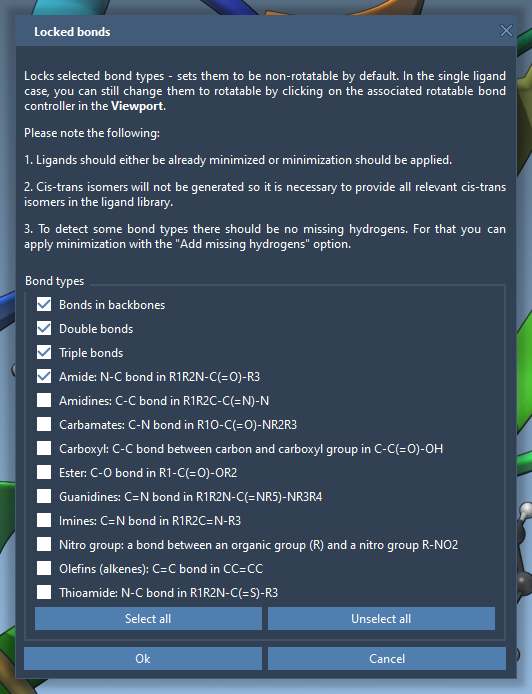
If adjusting each bond manually sounds tedious, AutoDock Vina Extended provides a way to automate this through the Locked bonds settings. Access this feature to specify certain bond types—such as amide or aromatic bonds—that should always remain non-rotatable for chemical consistency. Simply check the desired bond types in the dialog box and click OK.
Afterward, activate the Lock specific ligand bonds option to consistently apply these settings across your ligand or ligand library. This ensures important structural features are preserved while still permitting realistic flexibility.
Advanced Pre-Docking Preparation
Good docking practices often involve ensuring ligands are in the optimal conformations before starting the computation. If your ligands are in 2D or lack hydrogens, you can use the minimization option in AutoDock Vina Extended. This feature not only generates a realistic 3D geometry but also ensures polar hydrogens are present for accurately identifying hydrogen bonds (H-bonds) during docking.
In the minimization settings, you can set the maximum number of minimization steps and stopping criteria. This allows you to balance speed with accuracy, preparing ligands for docking without unnecessary computational overhead.

Considerations for Ligand Libraries
When working with a ligand library, all ligands are treated as fully flexible by default. Similar to single ligands, you can apply the Locked bonds settings to uniform bond types across the entire library. This is particularly helpful for large-scale screenings where you want to systematically control flexibility without manually editing each ligand.
Final Thoughts
Efficiently handling ligand flexibility during docking can significantly impact the success and reliability of your study. Whether you’re docking a single ligand or an extensive library, AutoDock Vina Extended in SAMSON gives you all the tools to tailor rotatability, locking specific bonds and even pre-minimizing ligands for optimal results.
For a detailed step-by-step guide, consult the official documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON today at https://www.samson-connect.net.