





For molecular modelers, balancing speed and accuracy while setting up models can be a challenge. Whether you are refining geometries, exploring relaxed states, or performing force analyses, the process can be daunting without the right tools. Luckily, the UMA Force Field in SAMSON makes configuring machine-learning-based simulations more accessible and efficient. In this post, we’ll discuss how to set up UMA simulations, making your molecular modeling pipeline simpler and faster.
The Setup Process, Simplified
UMA (Universal Models for Atoms) is well-suited for tasks such as fast geometry refinement and qualitative force analysis, but to unleash its potential, you need the right setup. Ensuring a smooth configuration will save time and enhance your modeling experience. Here’s how:
- Open the right document: Start by opening a SAMSON document that contains your molecular or material systems.
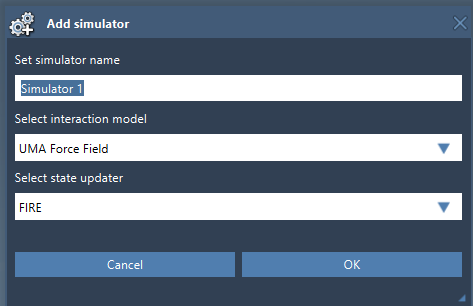
- Add a simulation: Use the menu path
Edit > Simulate > Add simulatorand select UMA Force Field in the interaction models list. - Configure the state updater: Choose a state updater like FIRE to define how your system evolves during the simulation.
- Start the UMA Setup: Once you’ve added a simulator, you’ll arrive at the UMA Force Field Setup window, where further configurations take place.
Choosing the Right Model and Task
In the setup window, you’ll need to make two foundational decisions:
- Select the model: UMA provides three machine-learning-based model options: UMA-S (fast workflow), UMA-S (lighter legacy), and UMA-M (large model, more capacity). Start with UMA-S for faster feedback unless you know you need UMA-M’s enhanced capacity.
- Select the task: Tasks are optimized for specific contexts:
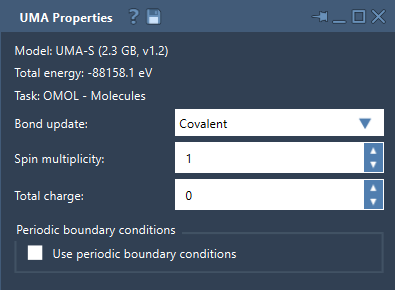
- OMOL – Molecules: Ideal for molecular systems. This is the only task requiring total charge and spin multiplicity.
- OMAT – Inorganic materials: For workflows involving bulk inorganic materials.
- ODAC – MOFs: Tailored for metal-organic frameworks and adsorption workflows.
- OC20 – Catalysis: Designed for catalytic surfaces and adsorbates.
- OMC – Molecular crystals: Best suited for molecular-crystal systems.
These customizable options ensure that the UMA Force Field aligns perfectly with your modeling objectives.
Tips for Getting Started
When configuring UMA, it’s essential to ensure that both the UMA Force Field and the Python Scripting extensions are installed and up to date. If it’s your first time using UMA, the platform will set up a dedicated Python environment, which may take time initially. Follow the on-screen instructions within SAMSON to ensure the installation is successful.
Pro Tip
If you’re unsure, begin with UMA-S for a faster workflow. You can always switch to UMA-M later if higher modeling capacities are required.
Ready to Run Your UMA Simulation?
Once the environment and model are ready, starting the simulation is straightforward. Use the menu path Edit > Simulate > Start, and UMA will begin evaluating your system’s energy and forces. During the simulation, you can refine structures interactively while leveraging UMA’s machine-learning capabilities for real-time updates.
At the start, energy values might not appear immediately in the Properties window; this is normal for the first evaluation pass. Once calculated, you’ll have interactive control over aspects like bond updates, periodic boundary conditions, and if applicable, charge and spin multiplicity for molecular systems.
Final Thoughts
The UMA Force Field in SAMSON streamlines molecular modeling setups by offering flexibility, speed, and precise configuration options tailored to various systems. For more in-depth instructions and advanced usage tips, check out the official documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at SAMSON Connect.