





When studying molecular interactions like ligand binding and unbinding, tracking the movement of specific atoms over time is often essential. Whether you’re preparing a reaction coordinate for free energy calculations or simply analyzing how a ligand navigates through a protein channel, you’ll likely need to export atomic trajectories in a structured format like PDB. If you use SAMSON, there’s a built-in tool that does just that.
In this post, we’ll walk through how to export only a subset of atoms—like a ligand—along a predefined path in SAMSON using the Export Along Paths extension. This is especially useful if you’ve calculated pathlines with the Ligand Path Finder and want to extract precise coordinates for downstream analysis or visualization.
Why Export a Subset of Atoms?
Exporting the full trajectory of all atoms in a system isn’t always necessary—or efficient. For instance:
- You might be interested in the motion of a small ligand only, not the entire protein-ligand complex.
- You may want clean input data for free energy profiling along a reaction path.
- You might be automating parameter generation for successive conformations in another tool or simulation workflow.
Step-by-step: Exporting a Ligand’s Path
In this example, we use a sample system of Lactose permease (PDB ID: 1PV7) and its ligand Thiodigalactosid (TDG), where two unbinding paths have already been generated.
- Open the Export Along Paths App
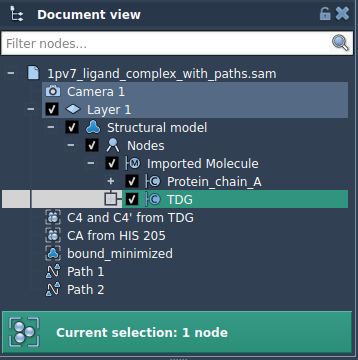
Launch it fromHome > Apps > All > Export Along Paths, or search it with Shift+E. - Select the Ligand
In the Document View, select the molecule subset you want to export—here, the ligand labeledTDG. - Add the Selection as a Model
Expand the Advanced panel in the app and click Add. This saves the current selection as a named group of atoms (called a model).
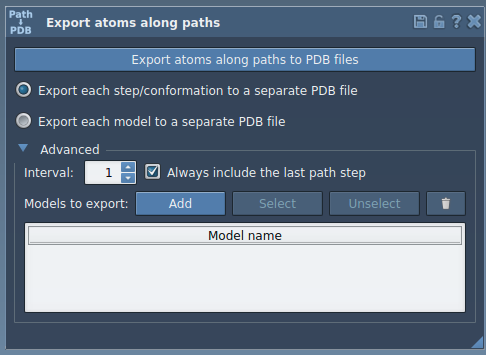
You can:
- Rename this model (helpful if you’re exporting multiple molecules).
- Add several sets of atoms if needed, e.g., the ligand and the binding site separately.
- Use the Select and Reset buttons to manage your models.
Export the Coordinates
- Choose how the frames are exported: either as one PDB containing all frames or a separate PDB file per frame.
- Select the path(s) you want—this determines the trajectory to follow.
- Click Export atoms along paths to PDB files. Select a destination folder and file name prefix when prompted.
This export is directly compatible with a number of analysis and visualization tools like VMD or PyMOL, or can be adapted into input for simulation packages such as GROMACS or AMBER.
Scenarios Where This Helps
- Extract ligand motions from exit paths.
- Convert optimized paths (e.g., from a nudged elastic band calculation) into coordinate snapshots.
- Study different atom groups independently, e.g., ligand vs backbone.
If this saves you time or helps in your research, feel free to share it with colleagues who might benefit too.
Learn more about the extension and the full workflow in the SAMSON documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.