





Understanding protein conformational changes can be both technically challenging and crucial for molecular modelers, especially in the context of drug design and protein-ligand interaction predictions. One such key conformational transformation is the opening movement of the SARS-CoV-2 spike protein—a process that determines whether the virus can bind to human cells and begin infection.
This transformation determines whether the receptor-binding domain (RBD) of the spike protein is in a ‘down’ (closed) position, where it cannot bind to human ACE2 receptors, or in an ‘up’ (open) position, where it’s accessible for binding. Modeling this transition requires not just structural snapshots, but a full trajectory that represents the motion in atomic detail.
The SAMSON platform makes it possible to simulate and visualize this transition. Using known PDB structures of the spike in its closed (6VXX) and open (6VYB) states, researchers can generate an interpolated trajectory of the opening process thanks to SAMSON’s ARAP and P-NEB modules. These animations help scientists visually understand the mechanics behind receptor recognition and viral entry.
Below is an example of the spike transitioning from its closed to open conformation, as seen from three different angles. These animations were calculated using ARAP and refined with the P-NEB module for improved motion realism:
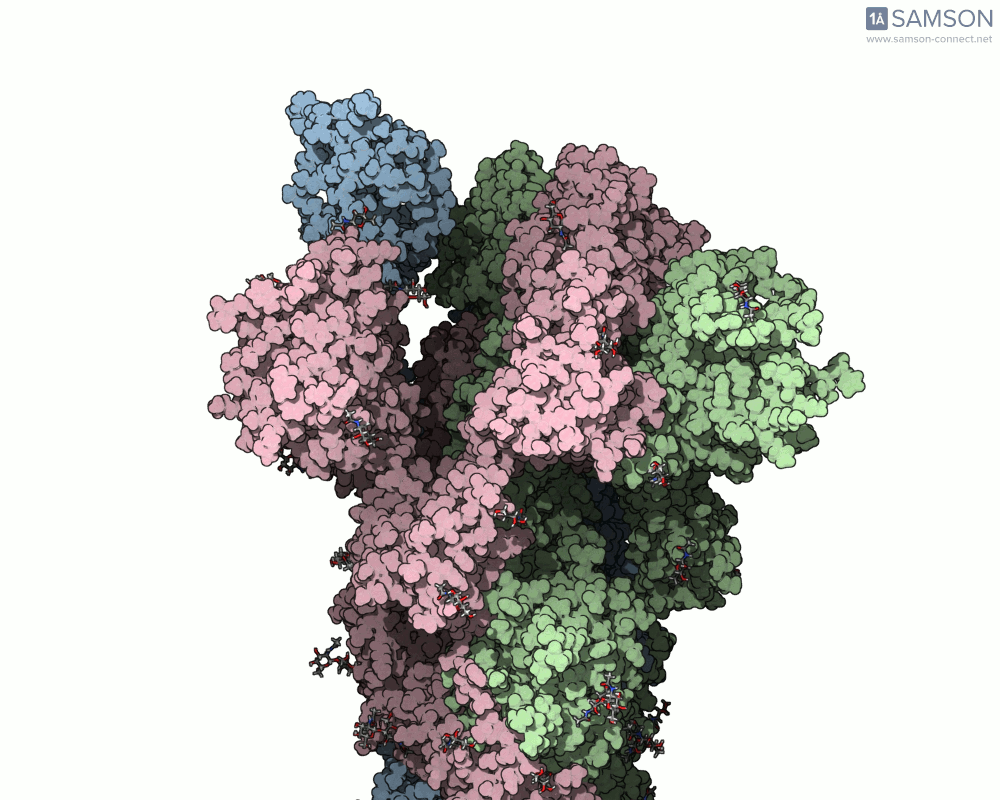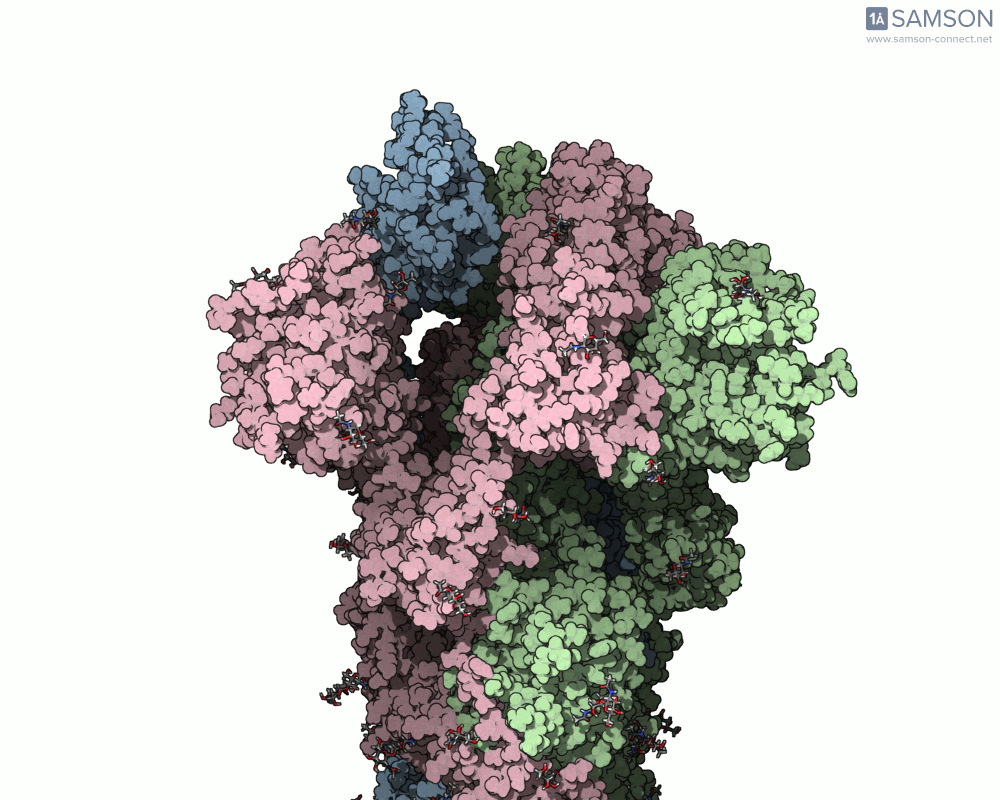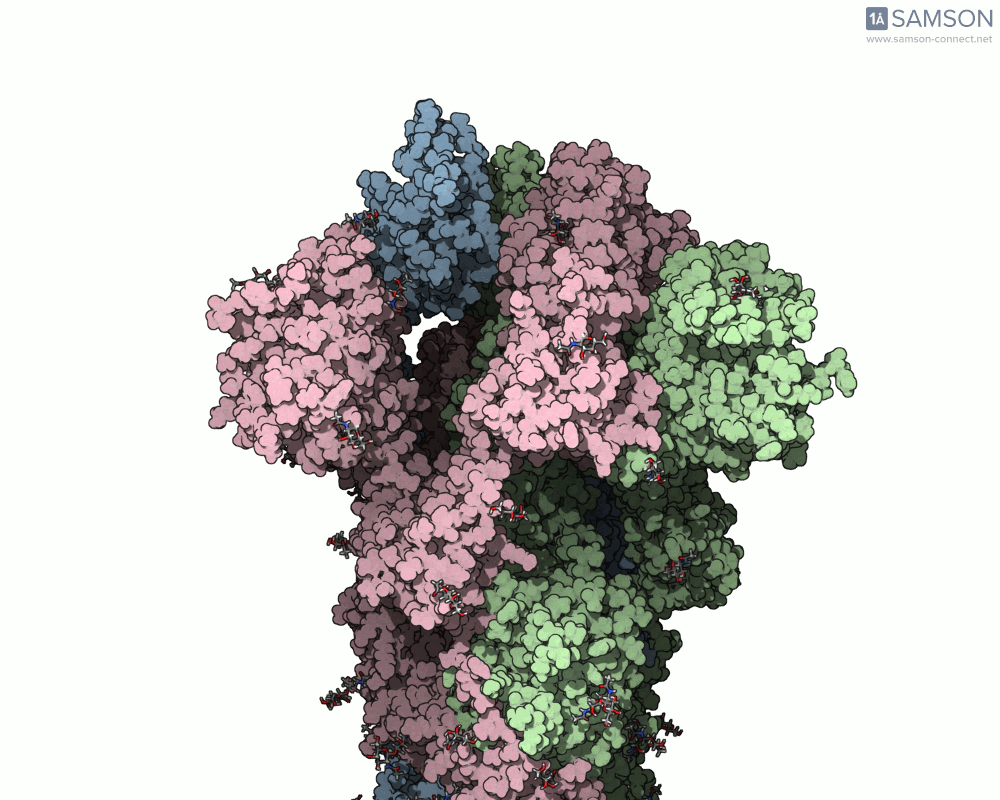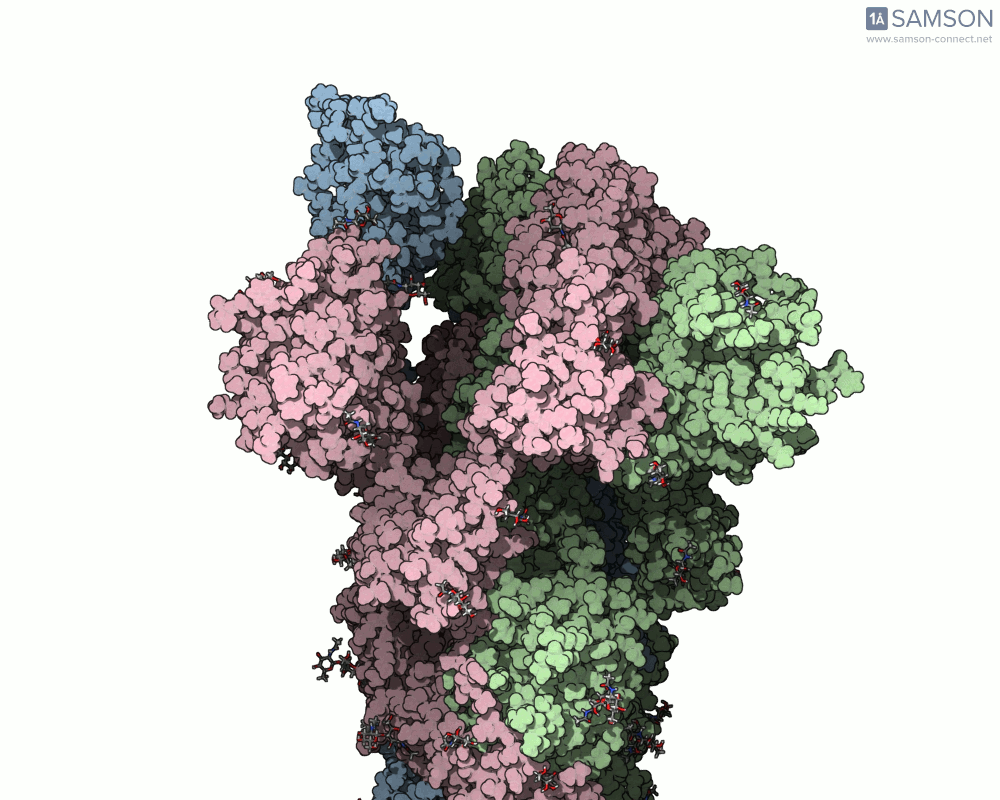
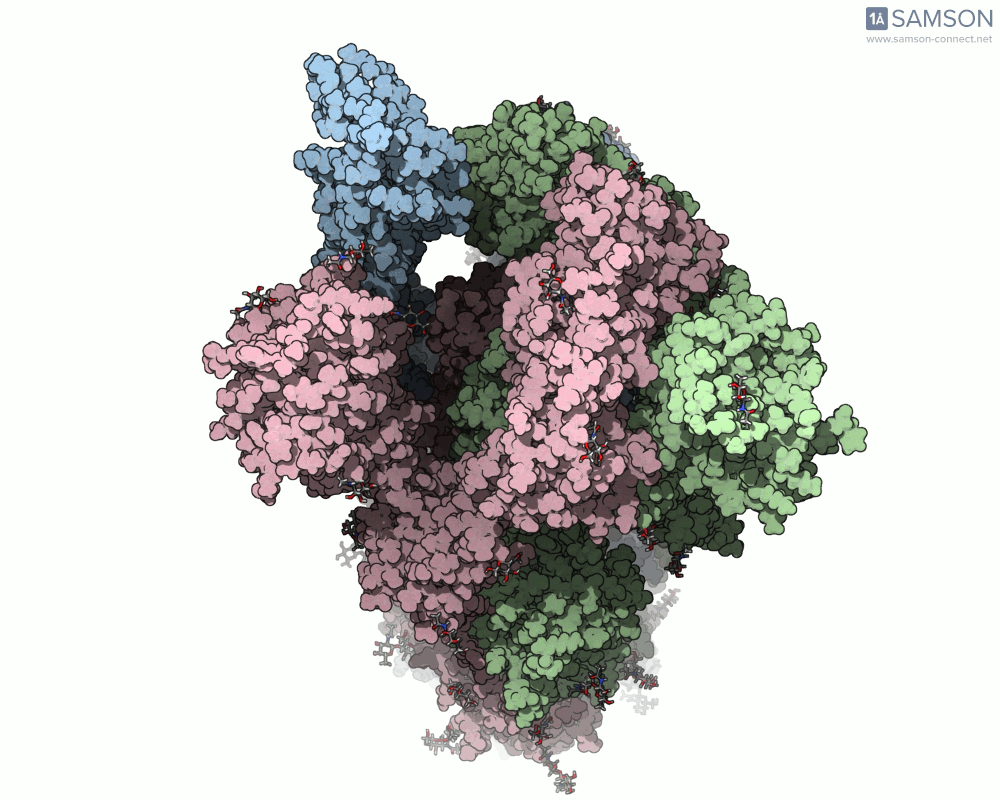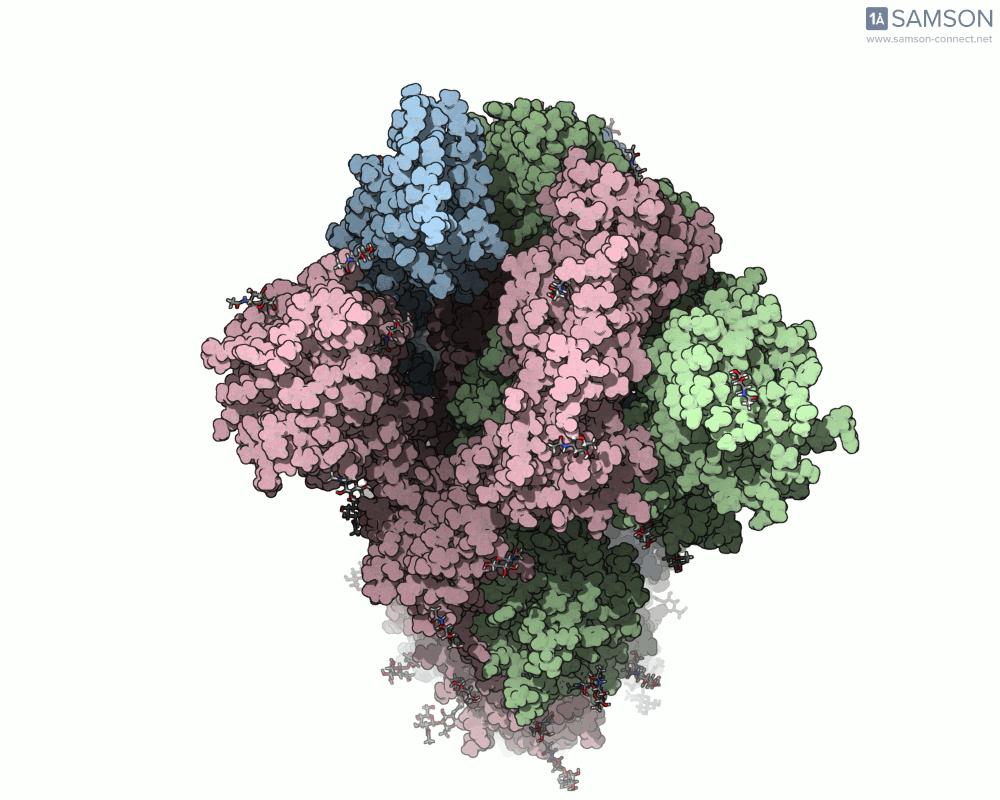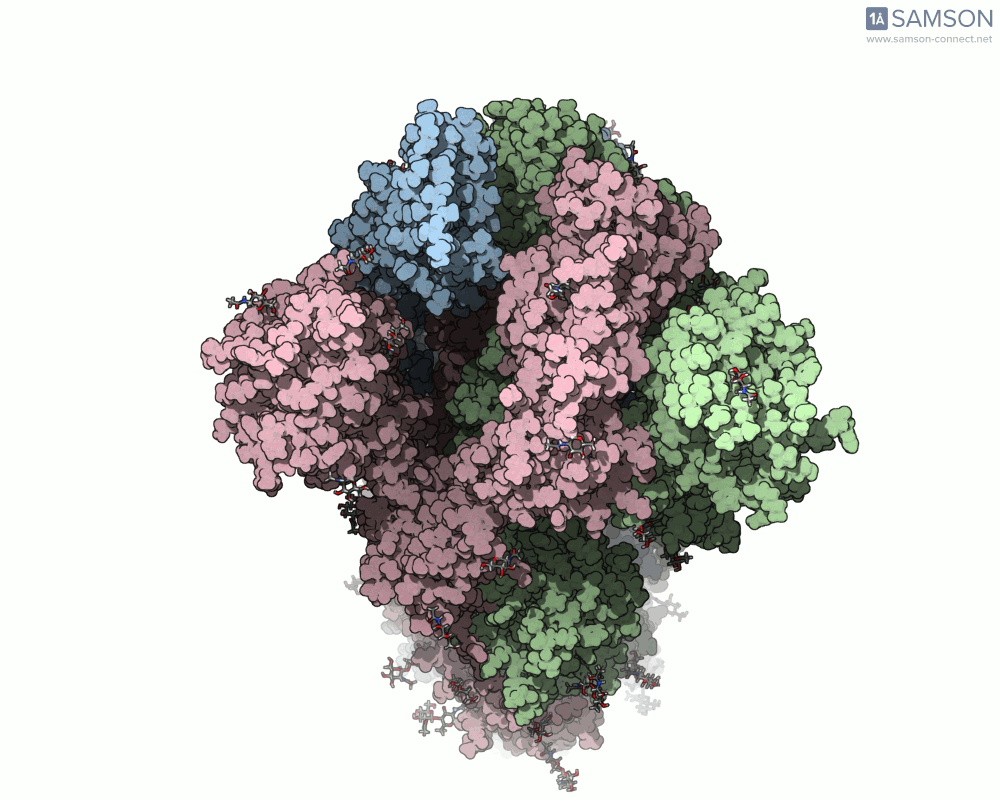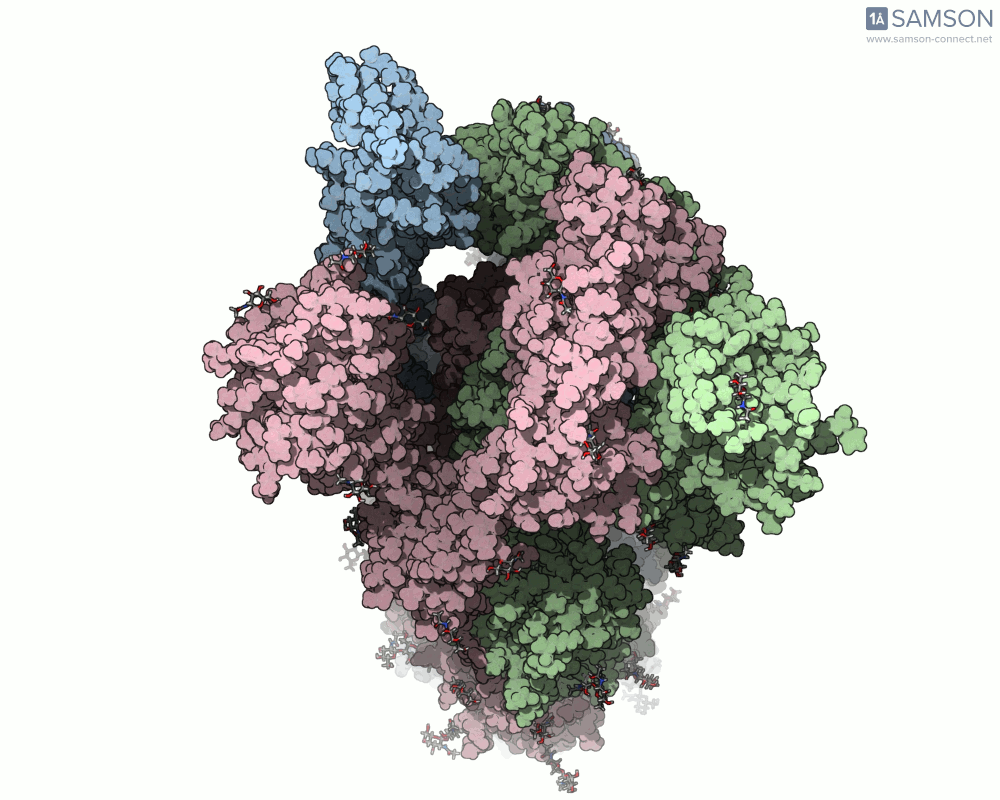
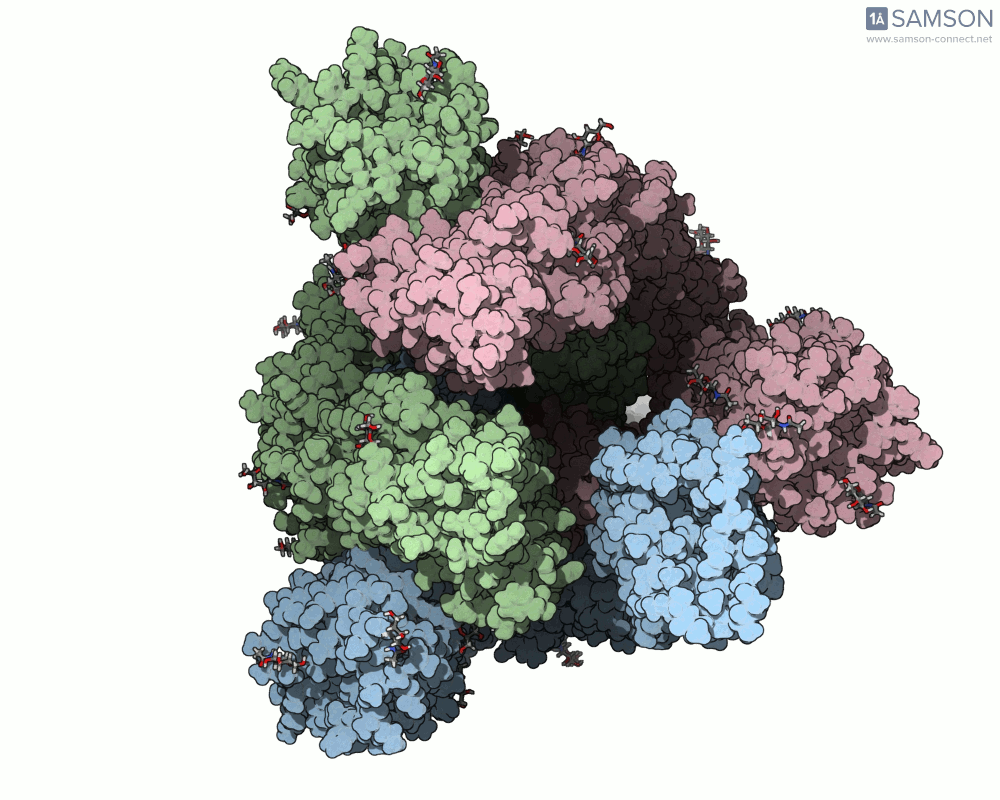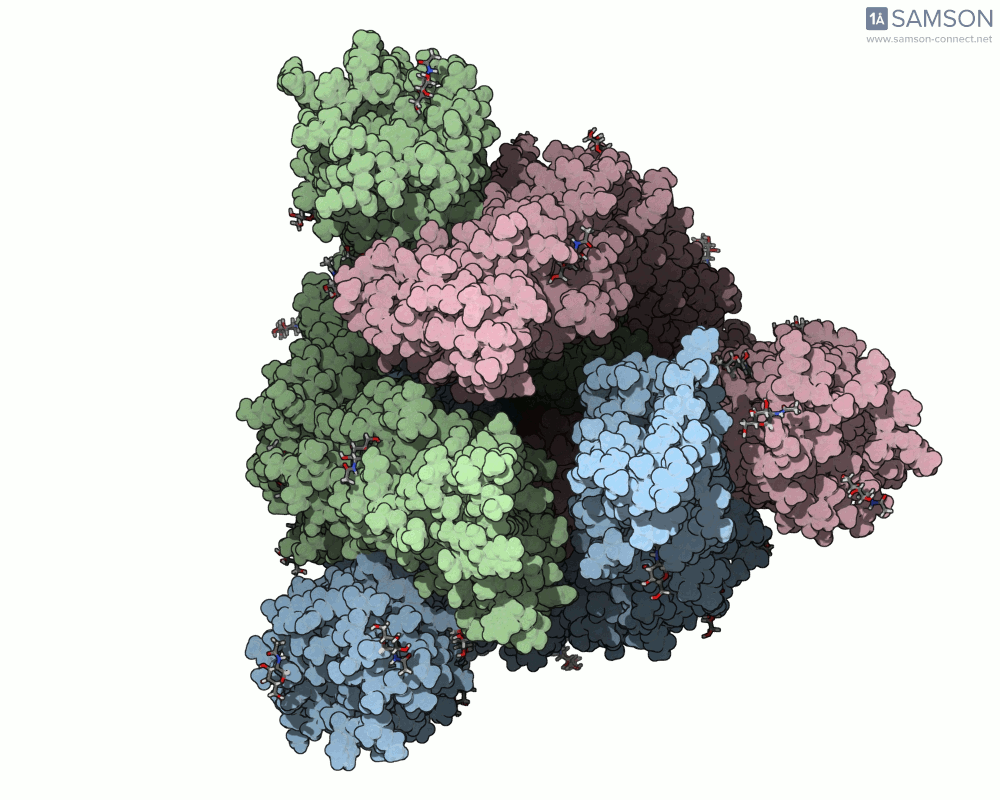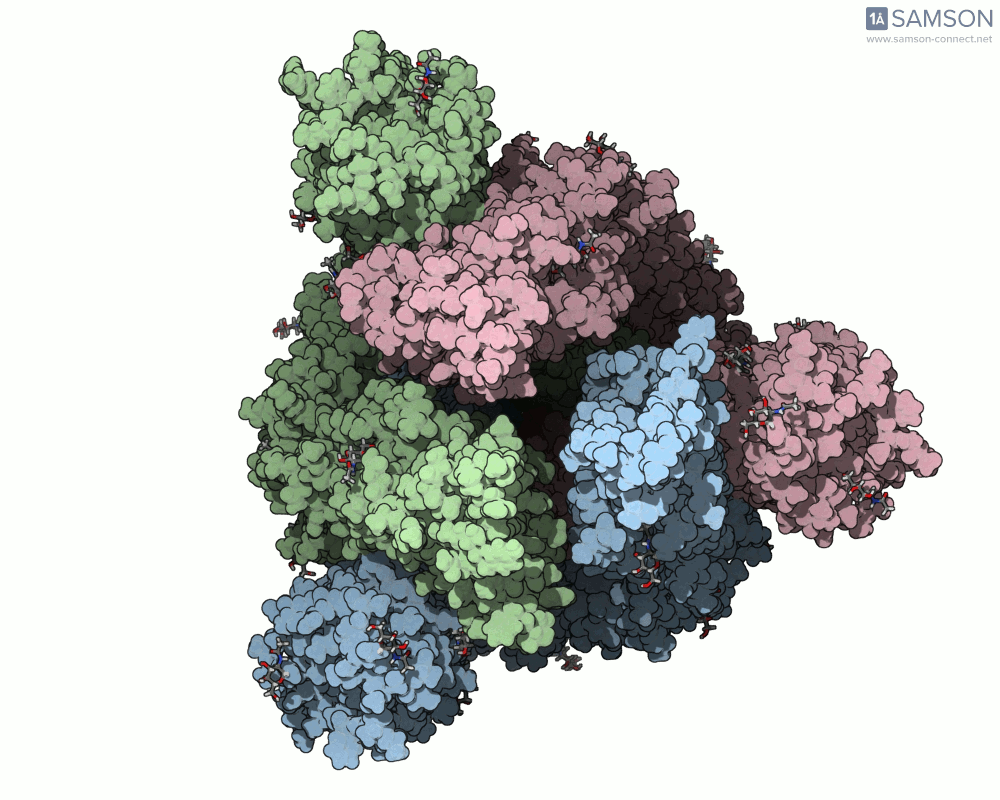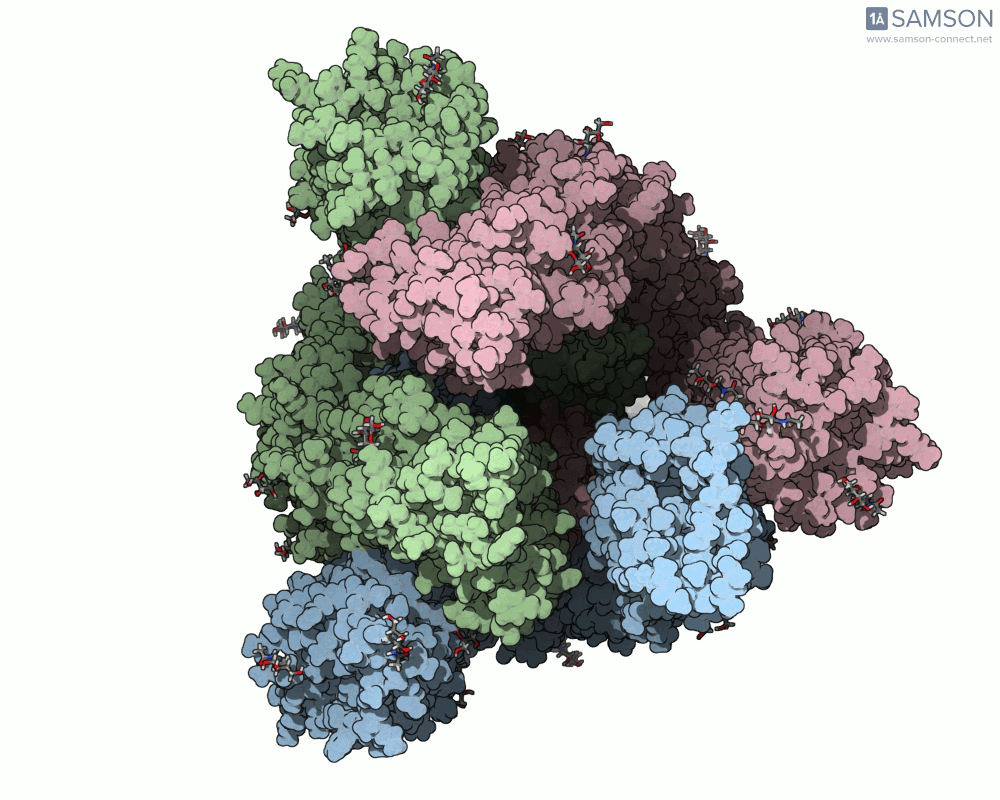
Why is this useful for molecular modelers?
- It provides visual and structural insight into a complex conformational shift relevant to viral infection mechanisms.
- Generated trajectories can be used as templates for docking experiments, especially for locating transition state inhibitors.
- The animation can guide where to focus when building antibodies or small molecules that disrupt the transition.
- Understanding motion also supports computational vaccine design via epitope exposure analysis.
All trajectory data shown here are available for free download in several common formats, including PDB trajectories and SAMSON files, which include open/close states, keyframes, and interpolated paths. These can be found on the original documentation page.
The calculations involve identifying two structural endpoints (open and closed), hydrogen addition and minimization, and path interpolation using ARAP before refining with P-NEB. The process takes only a few minutes and can be run on a standard laptop, making this a practical approach for research teams needing fast, transparent motion modeling without a cluster or GPU.
To learn more and explore the complete methodology or download the models and animations, visit the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.