





One of the most common bottlenecks in molecular design and cheminformatics is the need to systematically explore chemical space: how do structural modifications at specific molecular positions affect molecular properties like binding affinity or selectivity?
Instead of relying on scattered manual edits or external tools, the SMILES Manager extension in SAMSON provides an integrated solution to quickly define and search for chemical patterns directly from your working molecule—using the SMARTS language. This becomes especially useful in positional analogue scanning (PAS) workflows, where precise substitutions across multiple positions are needed.
In this post, we focus on a small yet powerful part of the PAS workflow in SAMSON: selecting a pattern to modify using SMARTS. We’ll walk through how to define a substructure of interest within your molecule—such as a hydrogenated aromatic carbon—and highlight where it appears in your molecule before generating analogs.
Why SMARTS Instead of Manual Selection?
SMARTS (Smiles Arbitrary Target Specification) is a querying language that allows for very specific chemical pattern definitions, from simple atom selections to elaborate fragments with constraints. Instead of pointing and clicking, you can leverage its expressivity to locate and select repeating features across a molecule that share the same chemical context.
Selecting Aromatic Carbons for Substitution
Let’s suppose your starting structure is the one considered in Pennington et al. with the SMILES code:
CN1C=C(C(=O)Nc2ccc(-c3ccccc3)c(c2)C(F)(F)F)C(=O)c2ccccc12
Using the SMILES Manager interface in SAMSON, you can input this SMILES directly or load it from a selection.
Finding Your Pattern
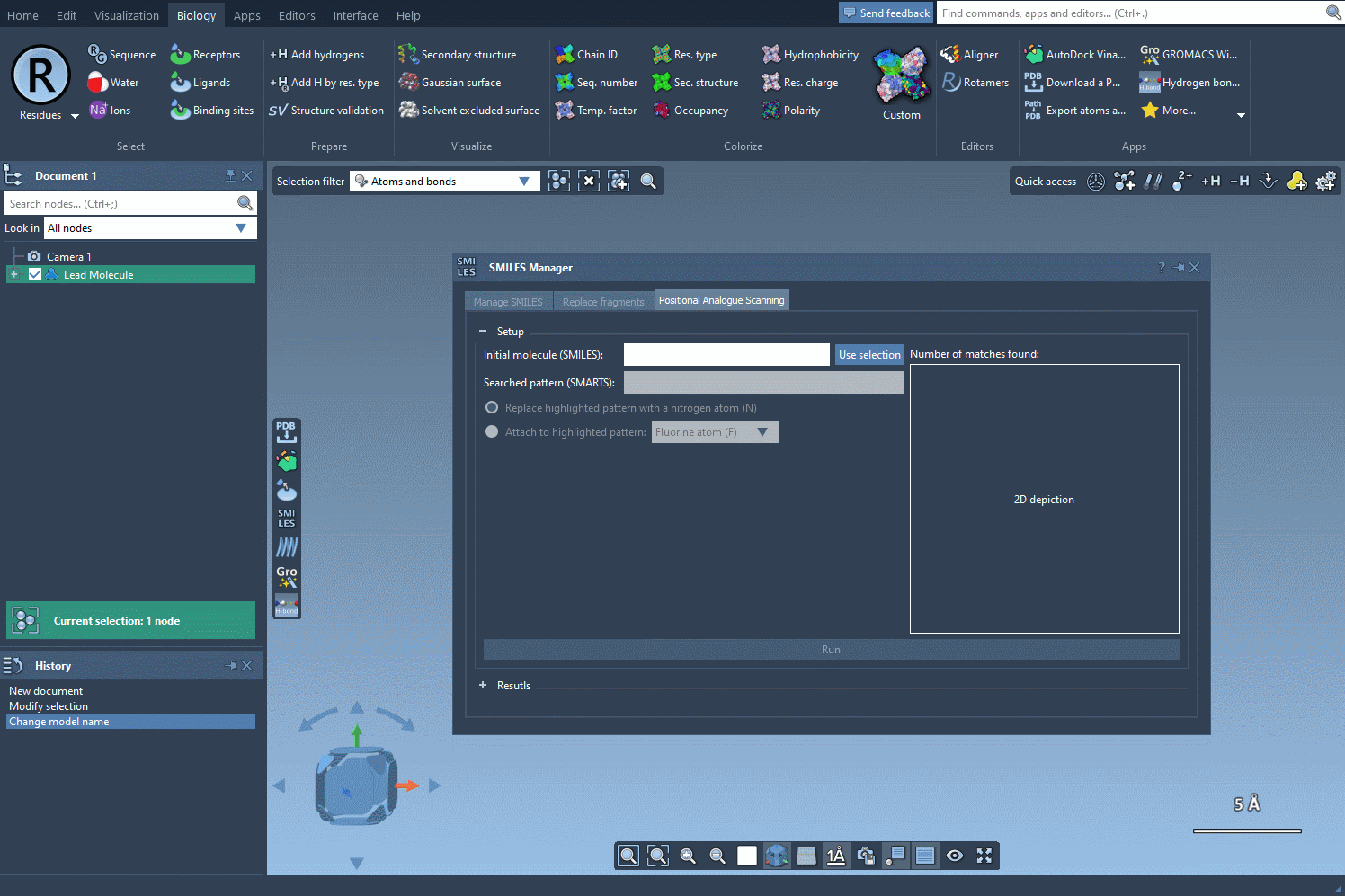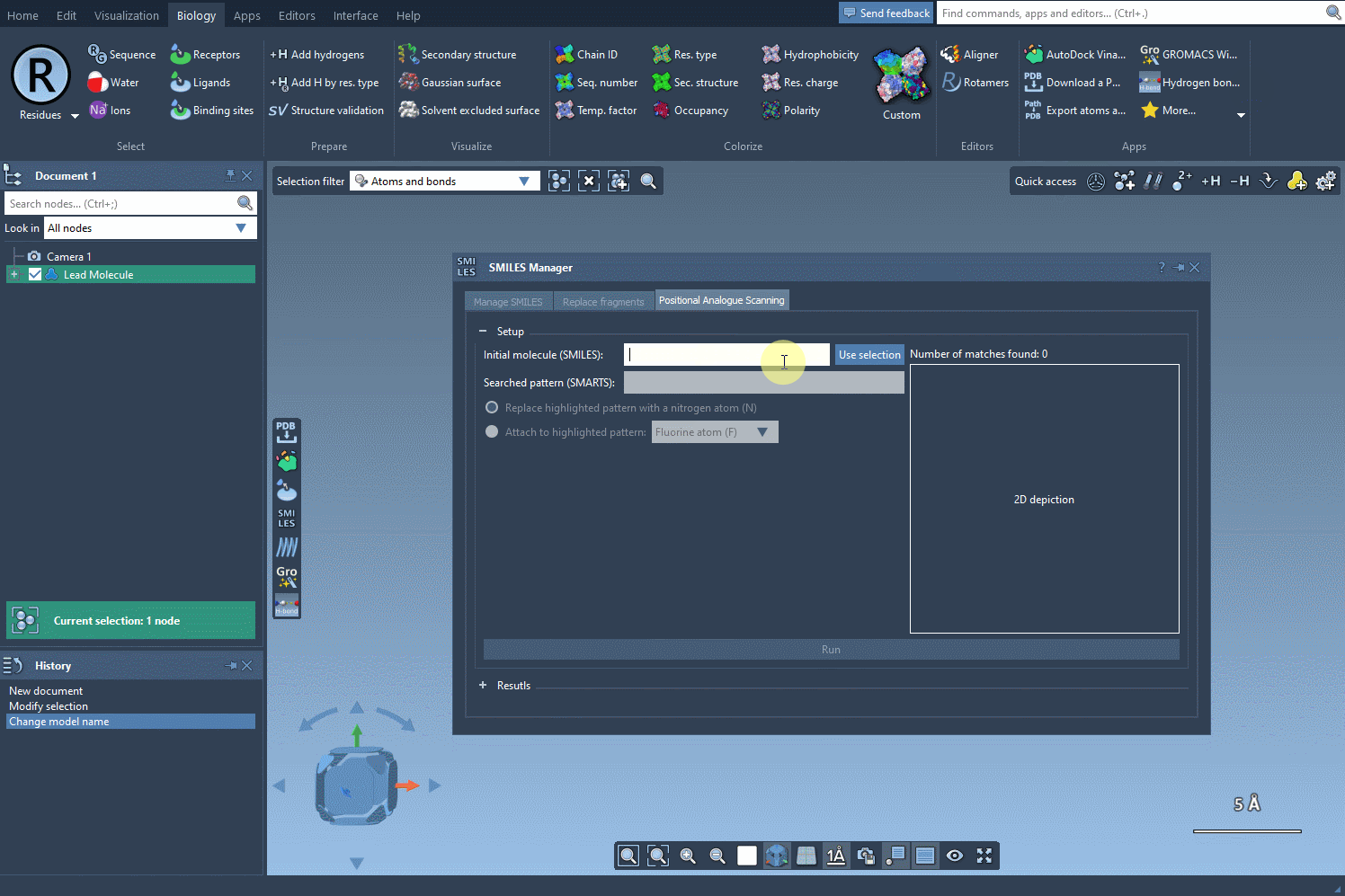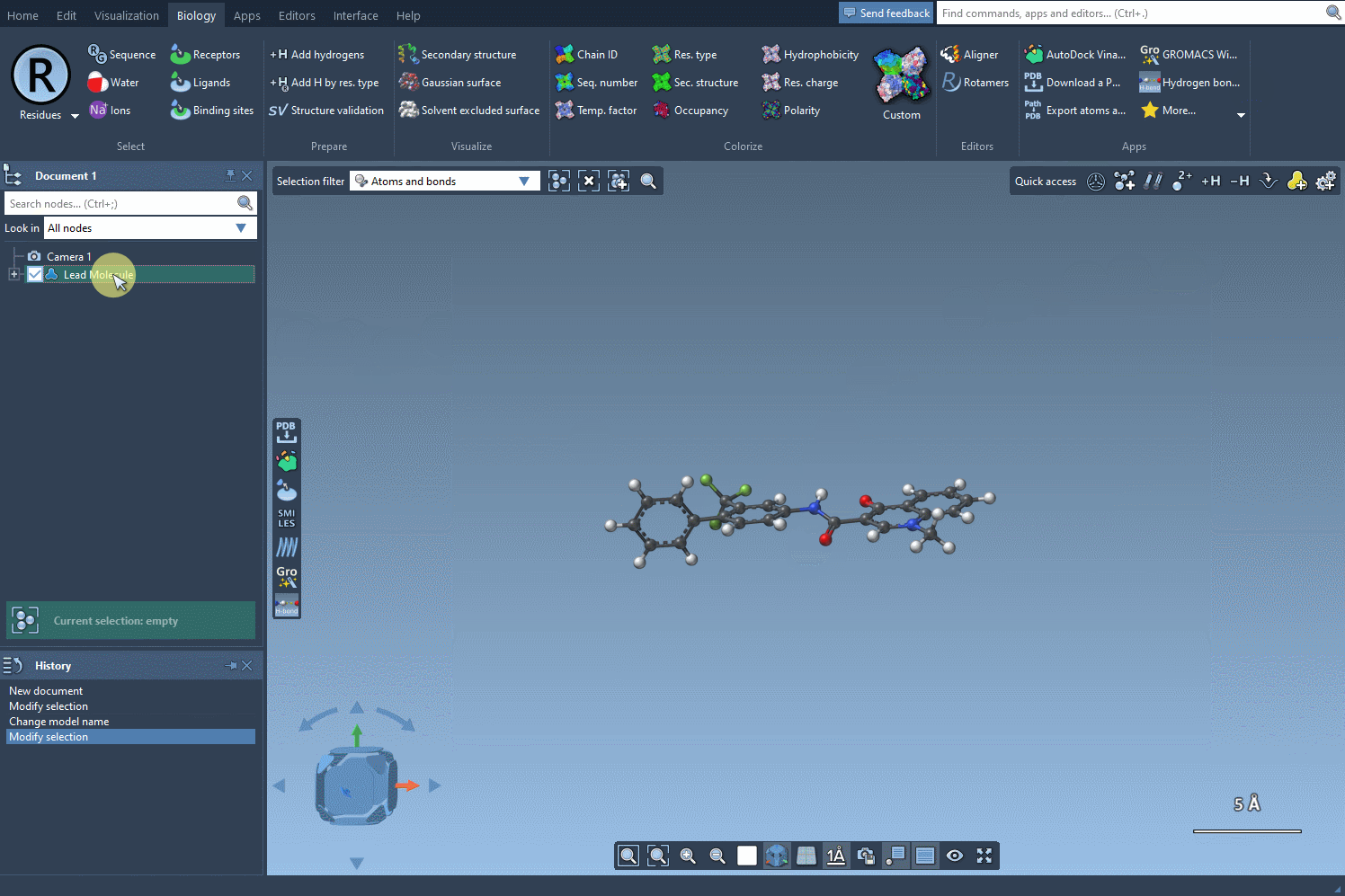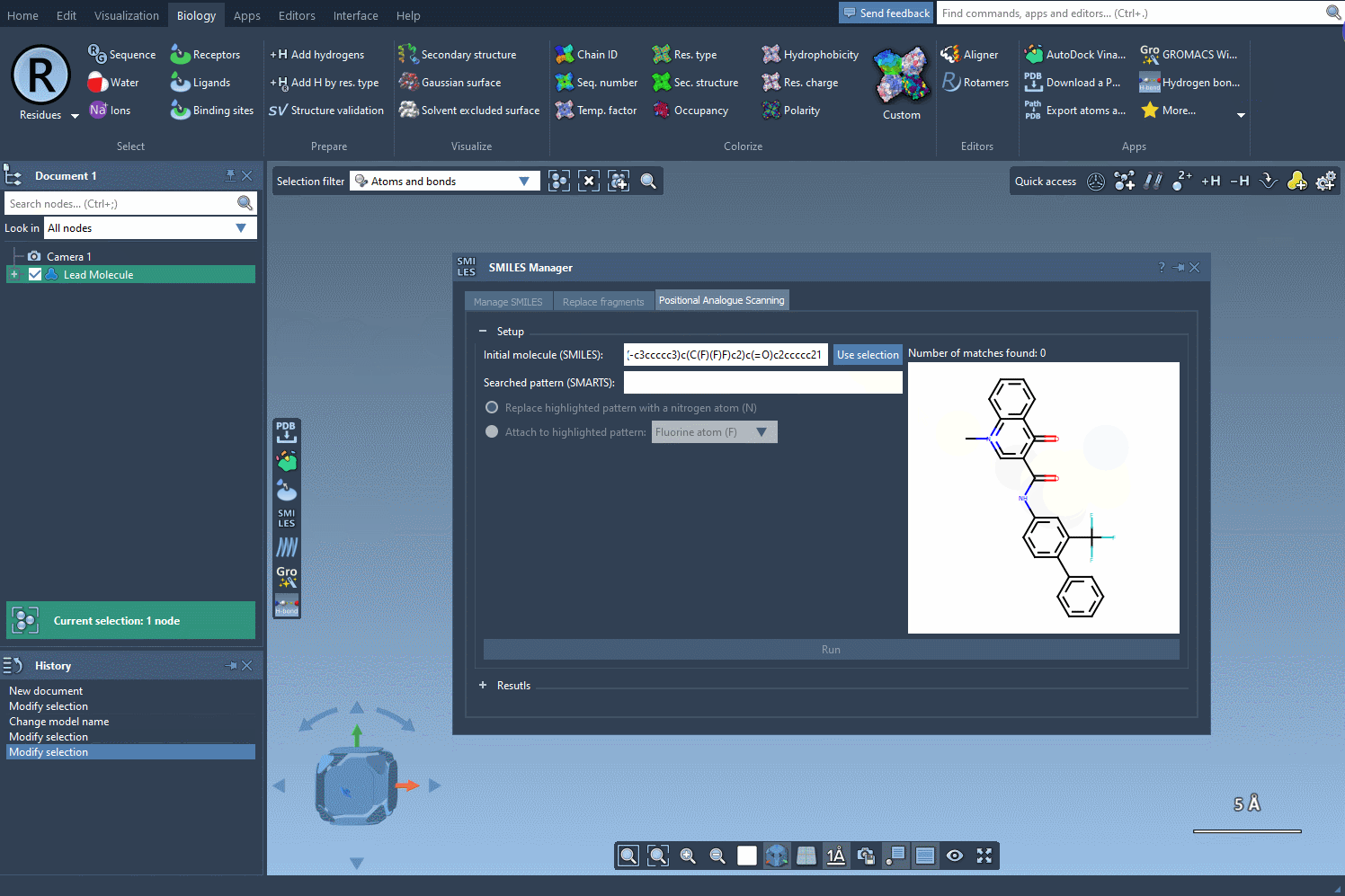
We want to find all aromatic carbons with a hydrogen (i.e., positions along the aromatic ring where we could substitute another group). To do this, we enter the SMARTS pattern [cH]—which captures hydrogenated aromatic carbon atoms—into the pattern search field within SMILES Manager.
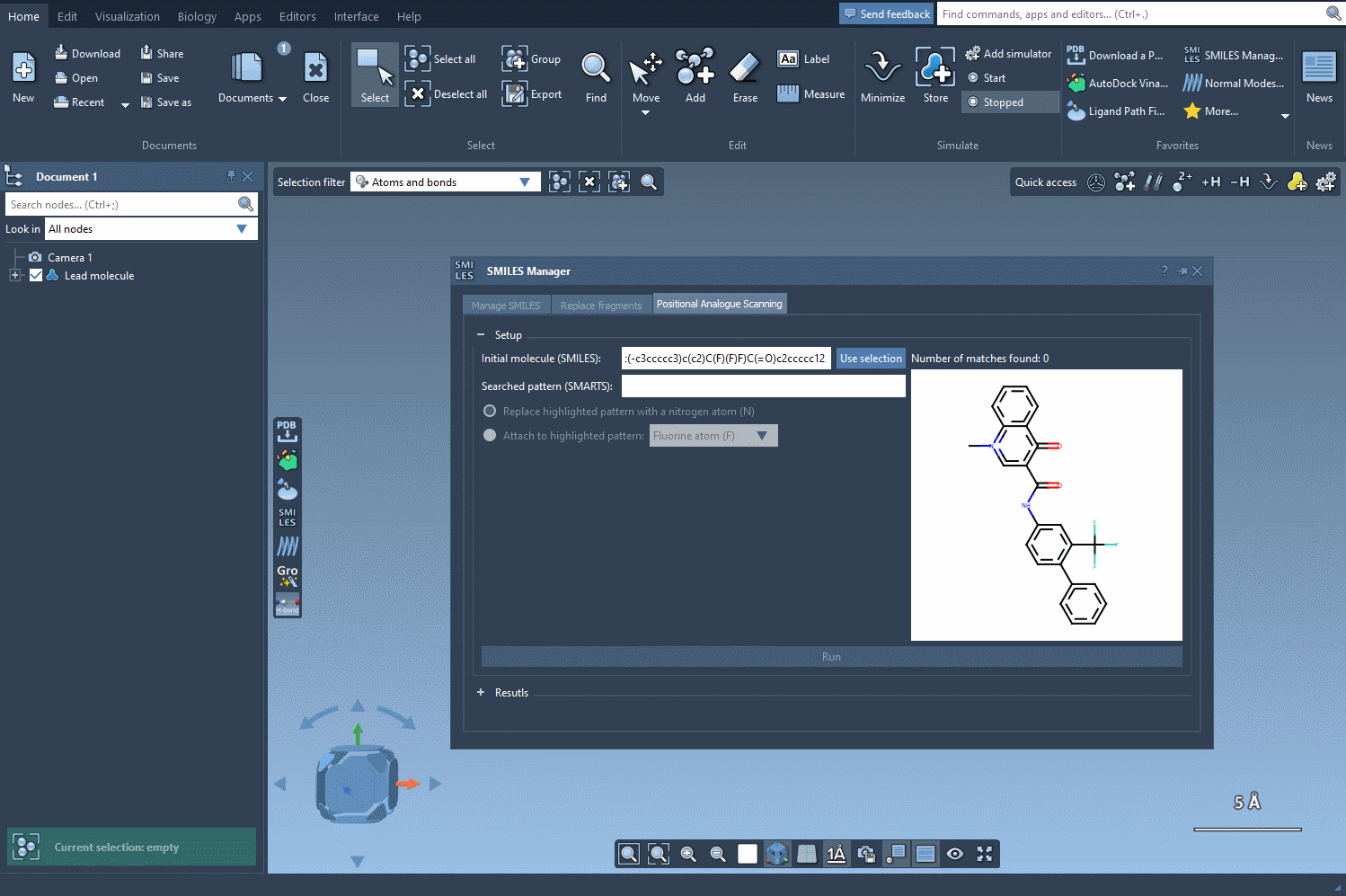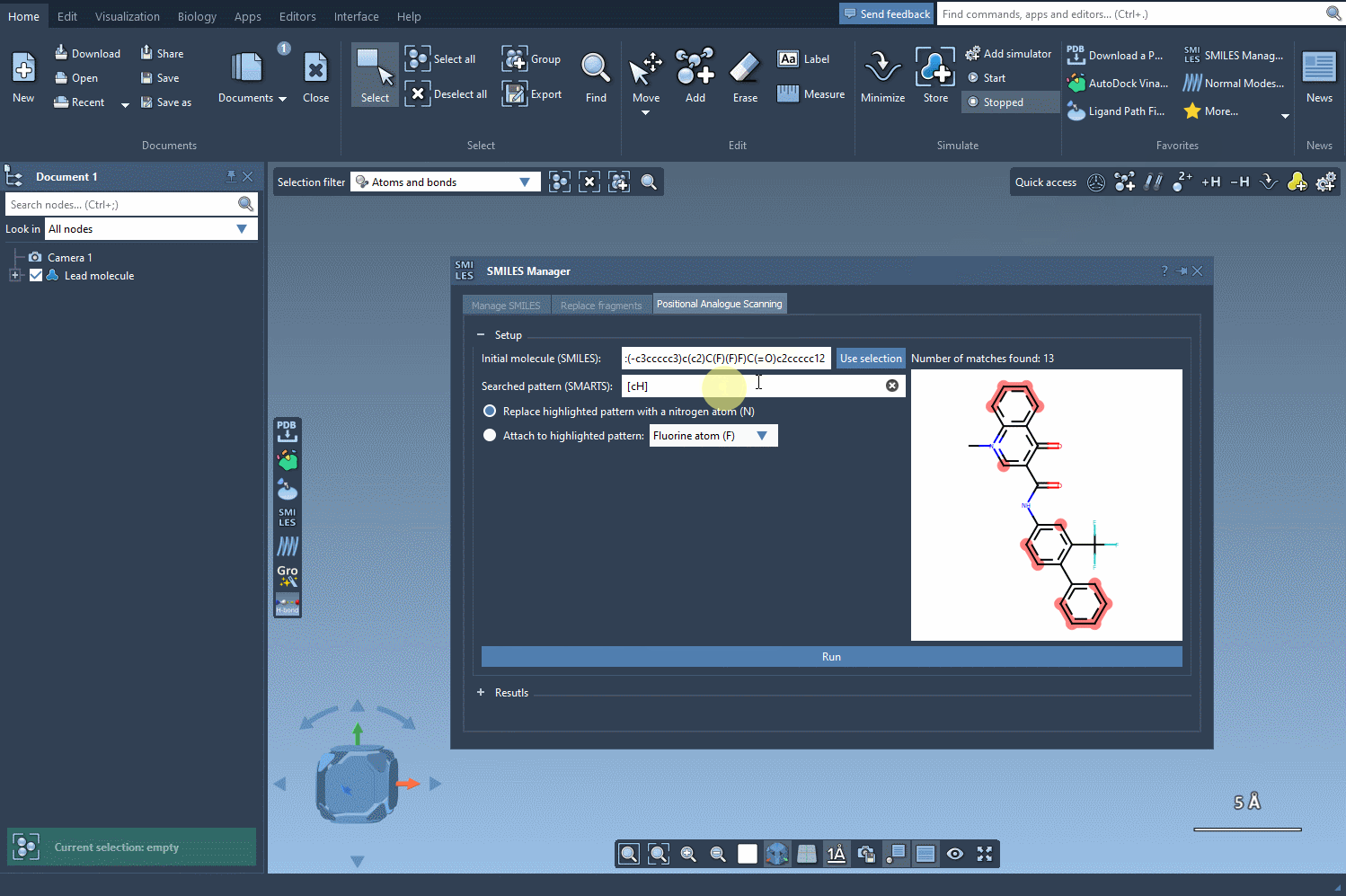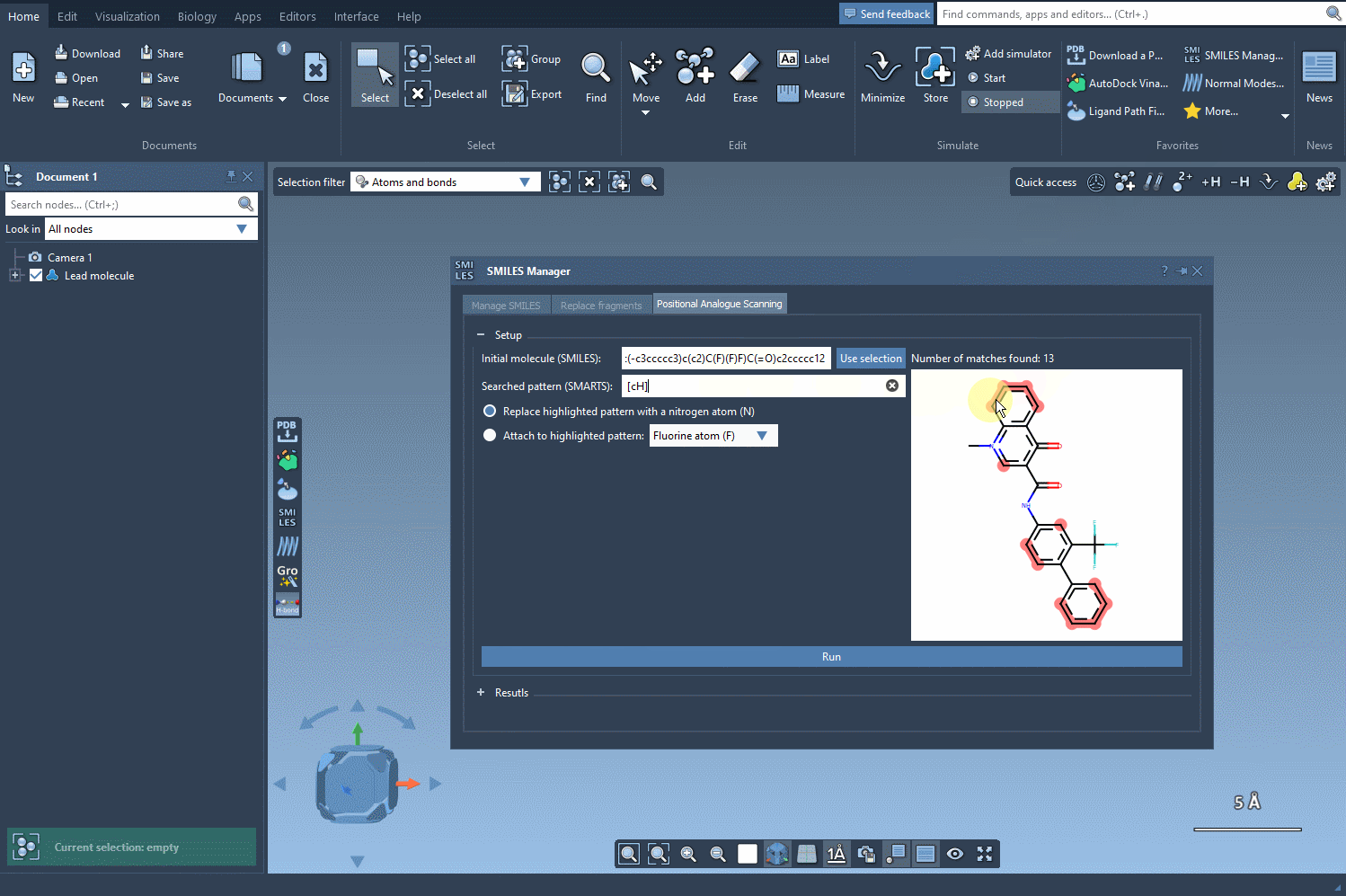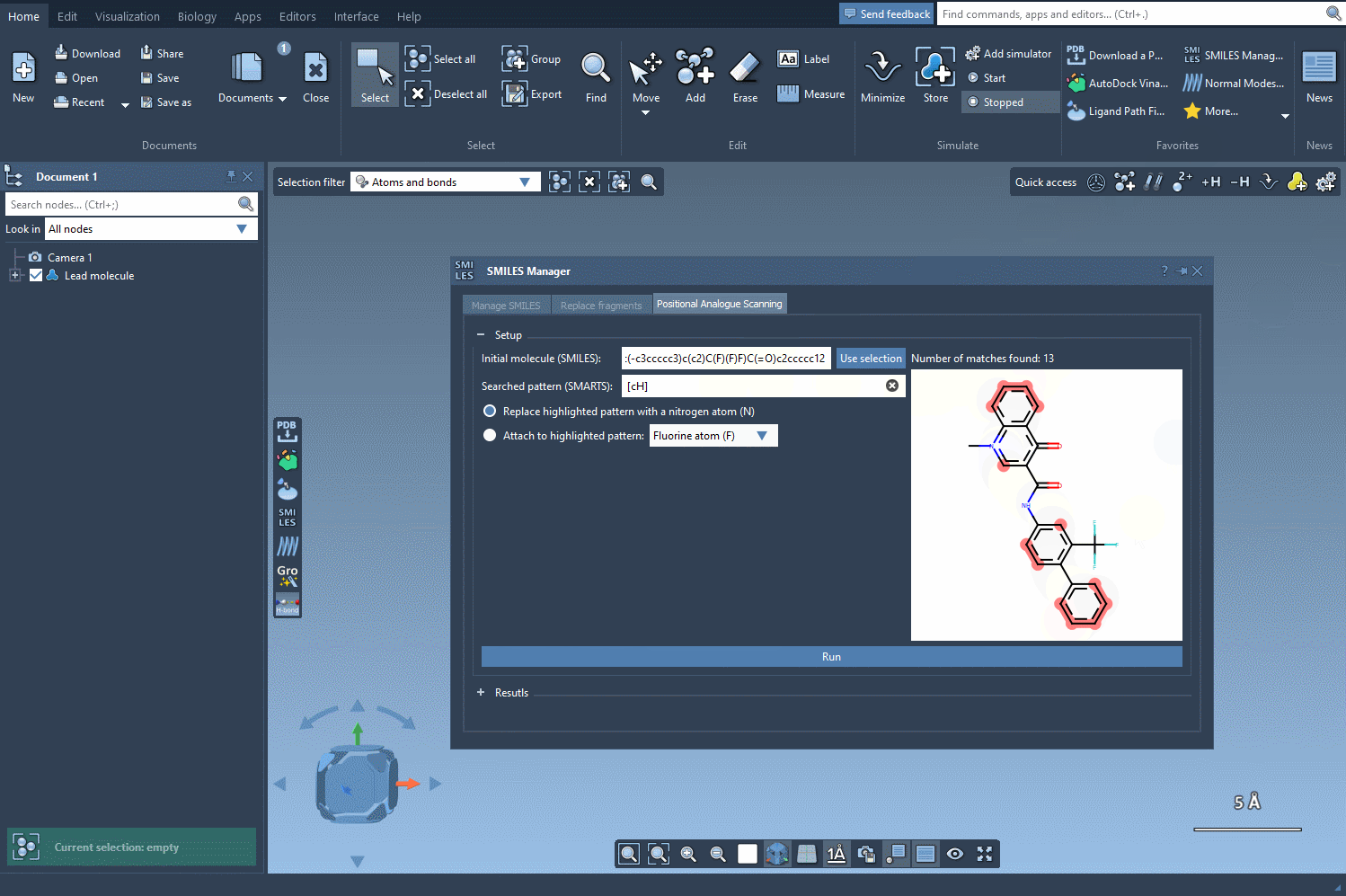
What’s helpful here is that when this SMARTS expression is applied, SAMSON highlights all matching atoms directly in the molecule. This visual feedback confirms which positions will be targeted in the generation of analogs, helping you avoid unwanted substitutions.
What Happens Next?
Once you’ve located your SMARTS-defined pattern, you can proceed to automatically generate molecular analogs by replacing the selected atoms (e.g., with a nitrogen) or by attaching other groups (like fluorine or methyl). From here, you can visualize results in both 2D and 3D, and evaluate binding via integrated docking tools.
Key Benefits
- No need to pre-process molecules or manually tag atoms—just use SMARTS directly in SAMSON.
- Visual verification ensures correct pattern matches before generating analogs.
- This step integrates seamlessly with other SMILES Manager features like analogue generation, structure visualization, and docking.
If you’ve been looking for a way to speed up and structure your analogue design workflow, try using SMARTS-based pattern detection in SMILES Manager—it’s fast, robust, and stays entirely within SAMSON’s interface.
Want to go further? Learn more in the full tutorial.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.