





Setting up a molecular mechanics simulation can feel like preparing for a journey with no map—especially when working with customizable force fields like the Universal Force Field (UFF). If you’ve ever hesitated because you weren’t sure how to initialize UFF or which choices impact your simulation most, this post is for you. Here, we go step-by-step through the setup process in SAMSON, aiming to remove ambiguity for those who want to use UFF accurately and efficiently.
Why UFF?
UFF (Universal Force Field) is highly versatile and supports elements across the periodic table, making it appealing for general-purpose simulations. Whether you’re modeling organic molecules, metals, or unusual chemistries, UFF can often provide a workable approximation. But setting it up correctly is key to usable results.
Step-by-Step: Setting Up UFF
Once you have a molecular system ready in SAMSON, follow these steps to add and configure UFF:
- Open your document containing the molecular system.
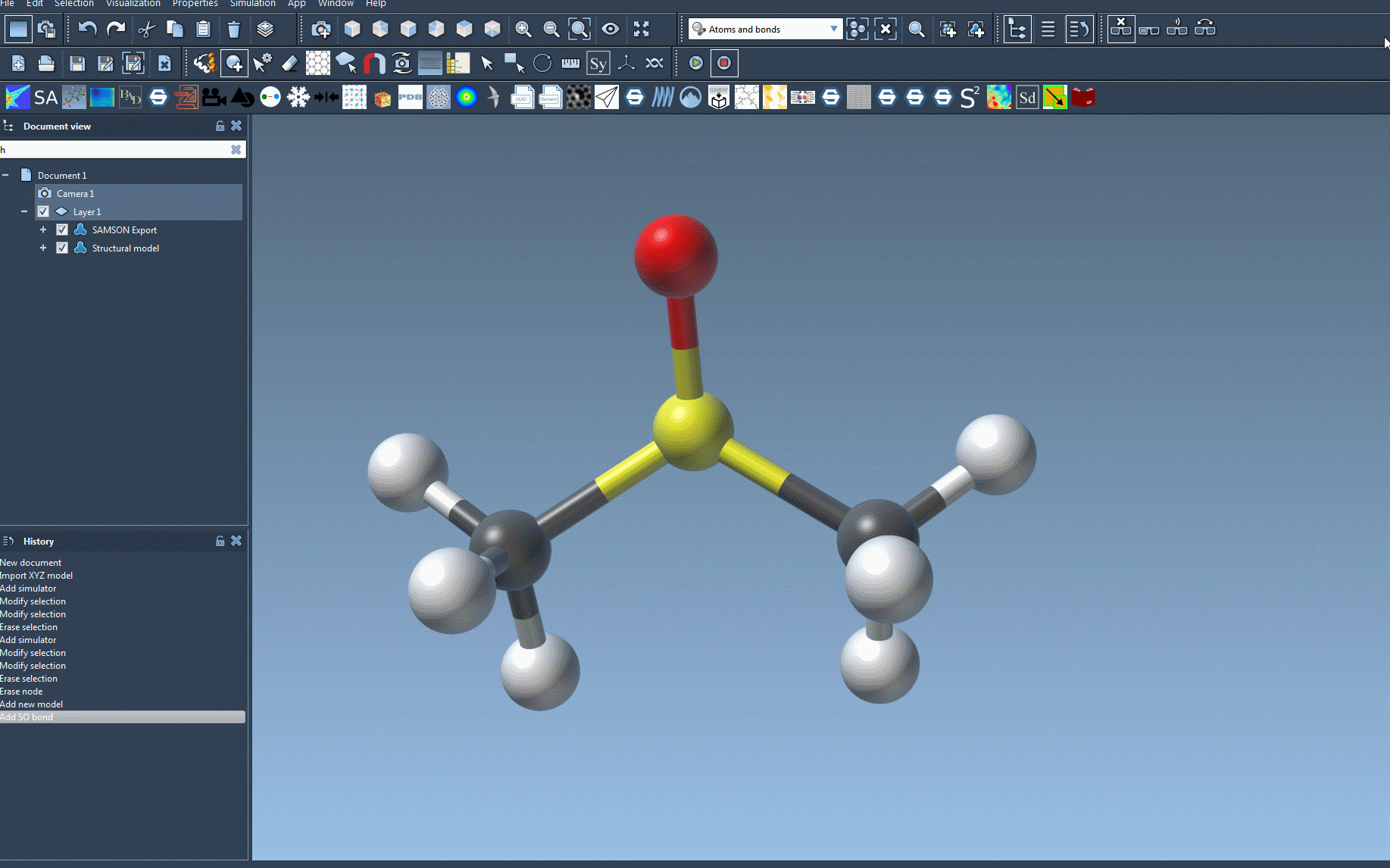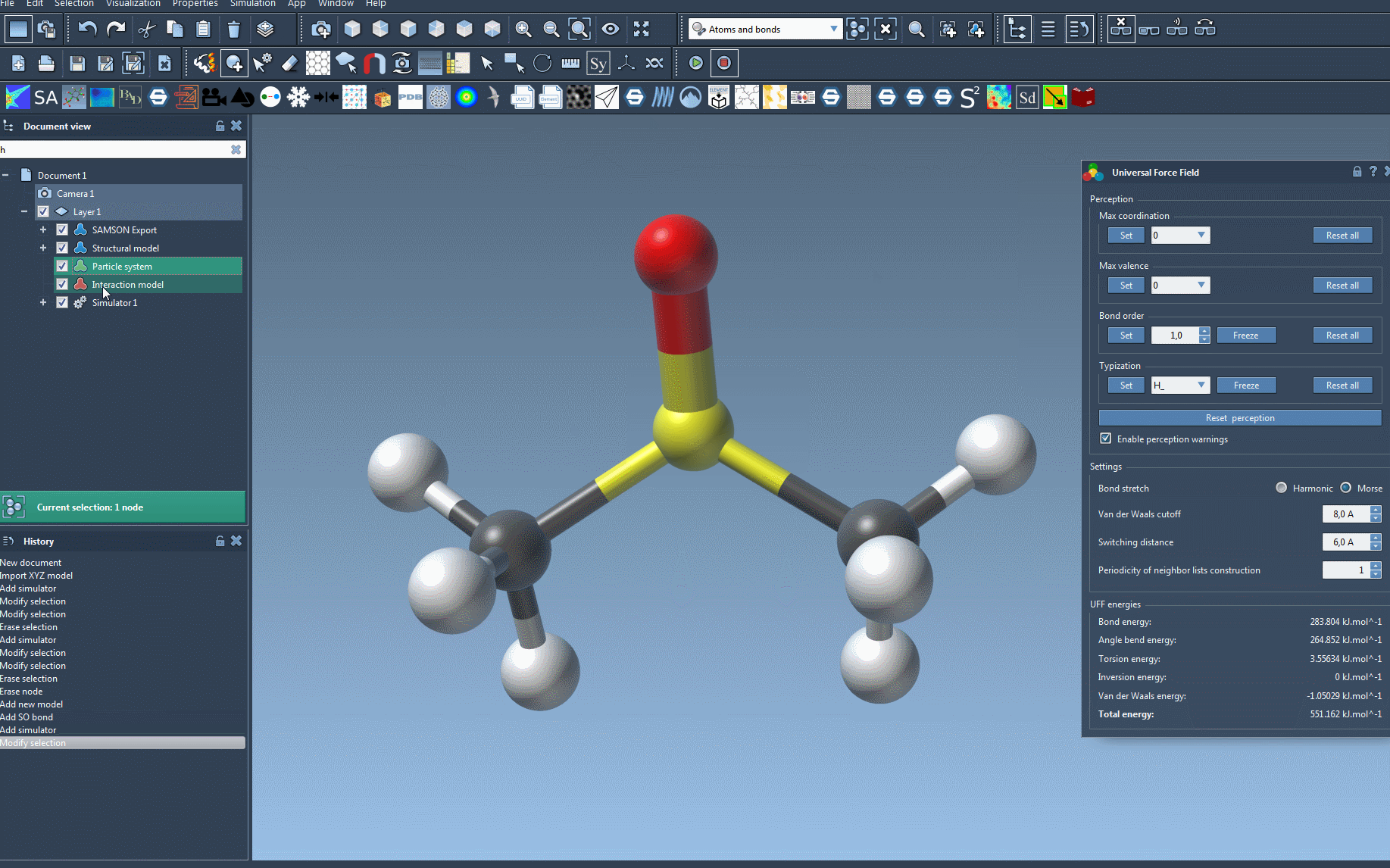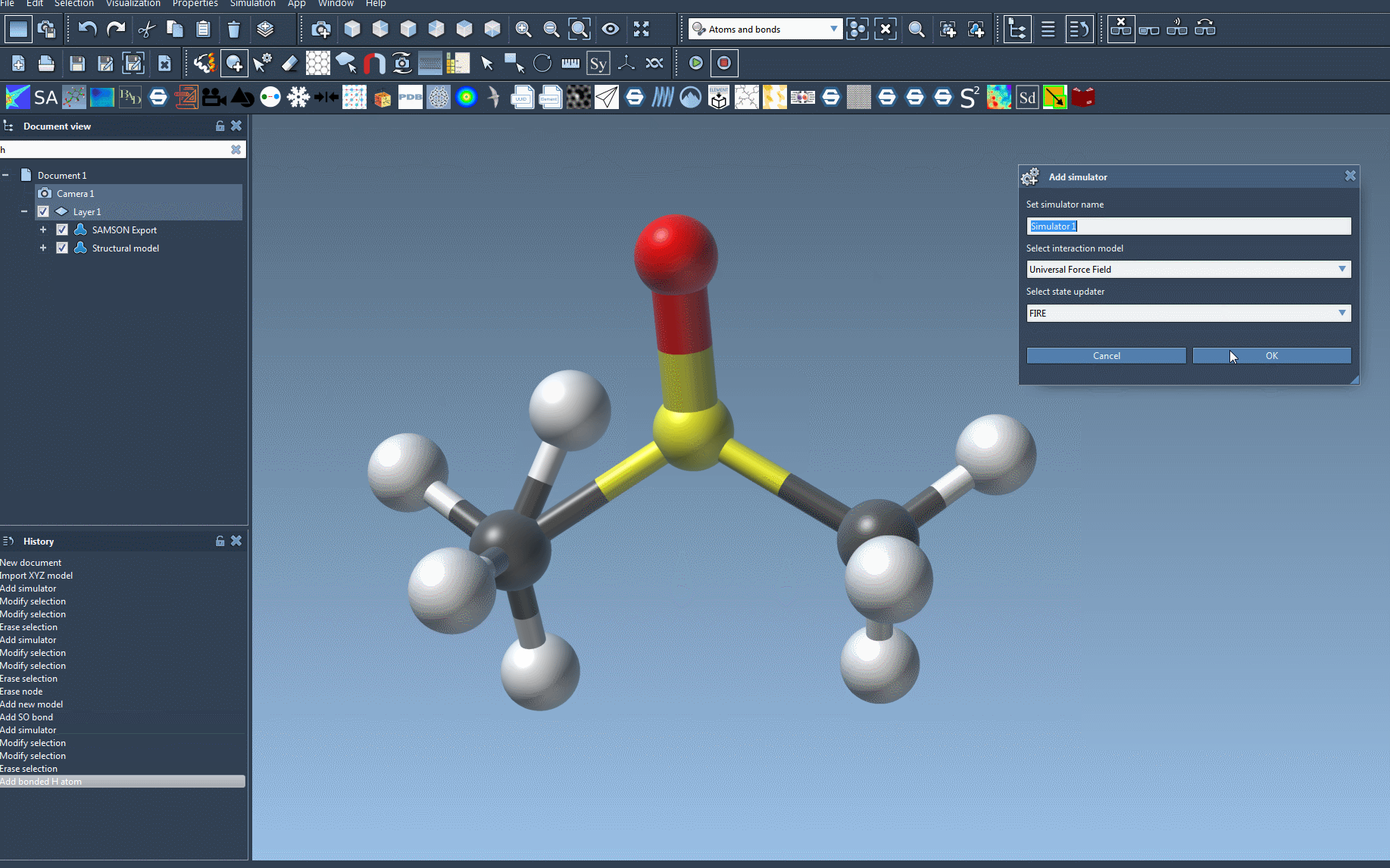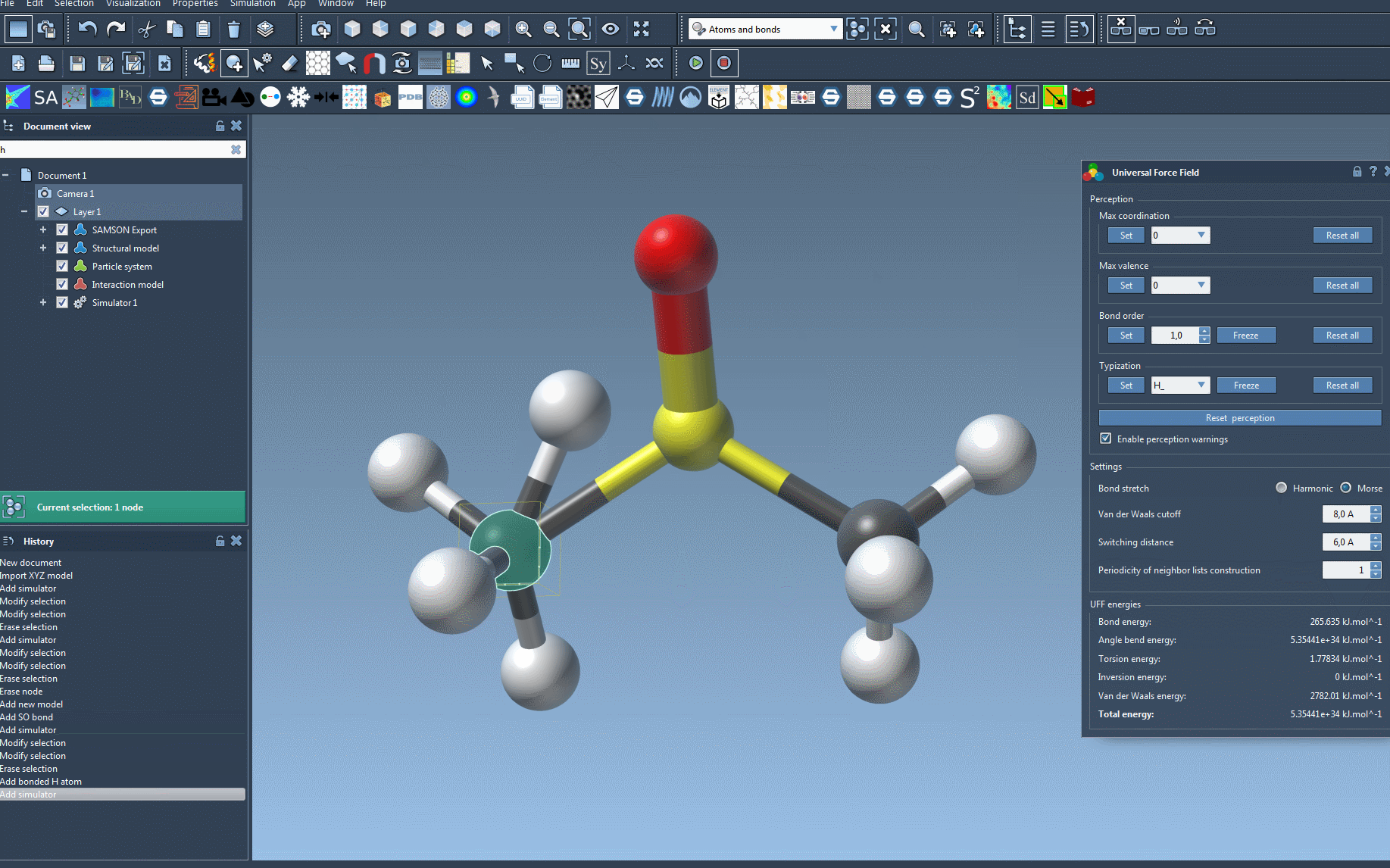
- Add a simulator: go to
Edit > Simulate > Add simulatoror pressCtrl + Shift + M(Cmd + Shift + Mon macOS). - Choose Universal Force Field from the list of interaction models.
- Select a state updater. One efficient option is FIRE (Fast Inertial Relaxation Engine).
- Click OK to proceed.
After this, the UFF setup window appears. Here’s where a couple of decisions matter:
- Use existing bonds: Check this if your system already has defined bonds and you want to preserve them as-is. Unchecked, SAMSON will recompute bonds based on atom types and positions.
- Click OK again to confirm.
What Happens Next?
The module now performs automatic molecular perception, which includes:
- Computing covalent bonds (if not already present).
- Determining bond orders.
- Assigning atom types for UFF.
Any inconsistencies—such as unexpected valences or ambiguous bonding—will be highlighted through alert messages. This early check can prevent issues later in simulation.
Tips for a Clean Setup
- When in doubt, try setting up your model with “Use existing bonds” unchecked, so you can leverage SAMSON’s automatic bond detection.
- If you notice warnings, inspect molecular geometry closely—common issues include overlapping atoms or incorrect connectivity.
Why Take the Time to Set This Up Properly?
Correct initialization with UFF ensures that you’re not simulating an unrealistic structure or spending time troubleshooting unstable behavior later. Especially when modeling complex or unfamiliar molecules, leveraging SAMSON’s perception scheme can save hours of manual edits.
Final Thought
One of the strengths of SAMSON’s UFF module is its flexibility. You can force bond orders, change atom types, or rely on automatic detection. But successful simulations usually begin with a clean setup using this guide. Accurate starting conditions give you the confidence your results are meaningful.
To learn more and explore other UFF features like custom typization or parameter tweaks, visit the full documentation: https://documentation.samson-connect.net/tutorials/uff/uff/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.