





For many molecular modelers and chemoinformaticians, turning a list of SMILES strings into meaningful 3D molecular structures remains a recurring task. Whether you’re preparing a virtual screening library, visualizing molecular conformations, or setting up a simulation, the process often involves multiple tools and manual effort.
This blog post shows how SAMSON’s SMILES Manager extension streamlines the conversion of SMILES into 2D and 3D structures, making the process fast and interactive. This can be a productivity booster for researchers who want to avoid scripting or repetitive tool switching.
Importing SMILES data
The SMILES Manager supports RDKit-style .smi and .txt files. You can import these via the File > Open menu, and the content will be populated in a table. For example, a SMILES file may look like this:
|
1 2 3 4 |
SMILES Name COc1cc(CNS(=O)(=O)CCCCC=CC(C)C)ccc1O Mol1 COc1cc(CNC(CCCCC=CC(C)C)C(F)(F)F)ccc1O Mol2 |
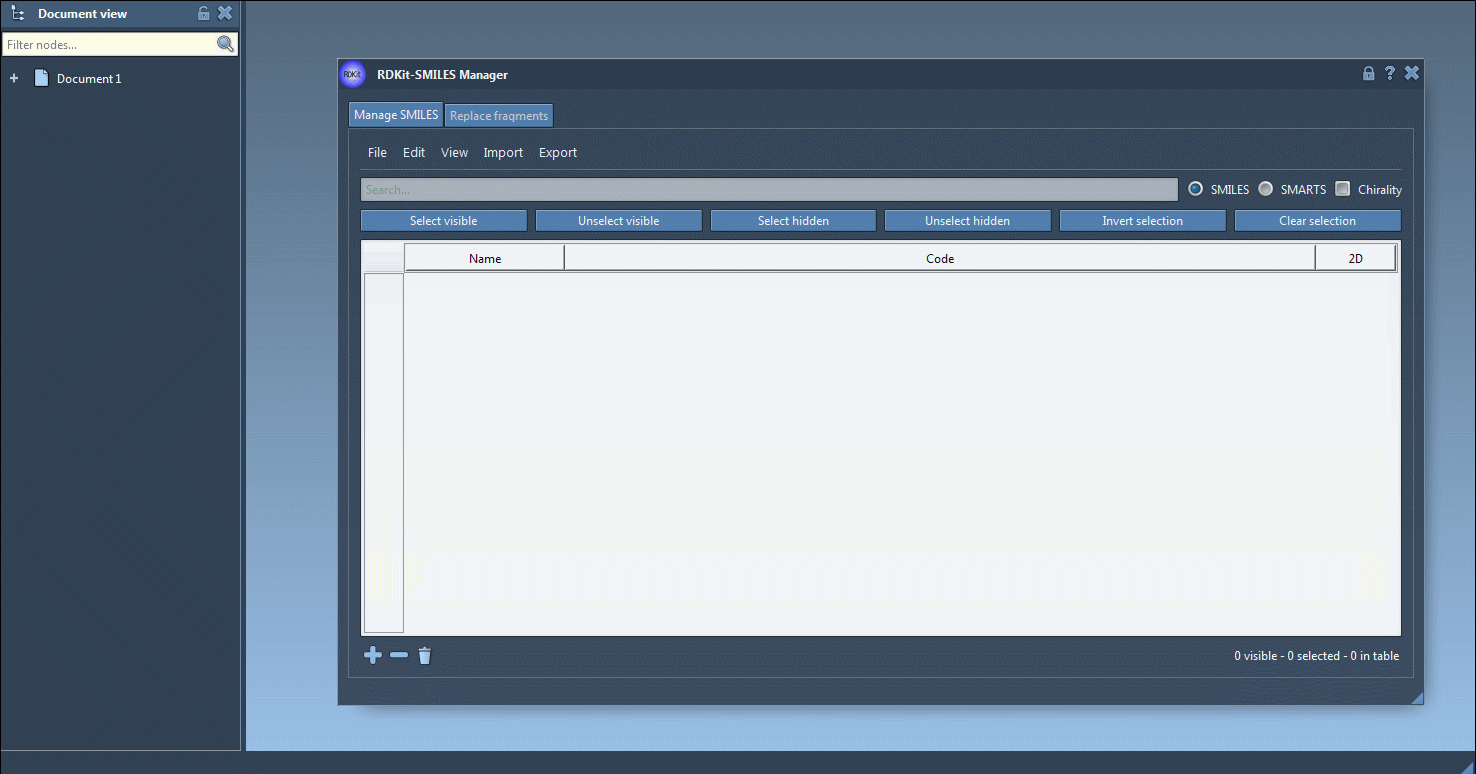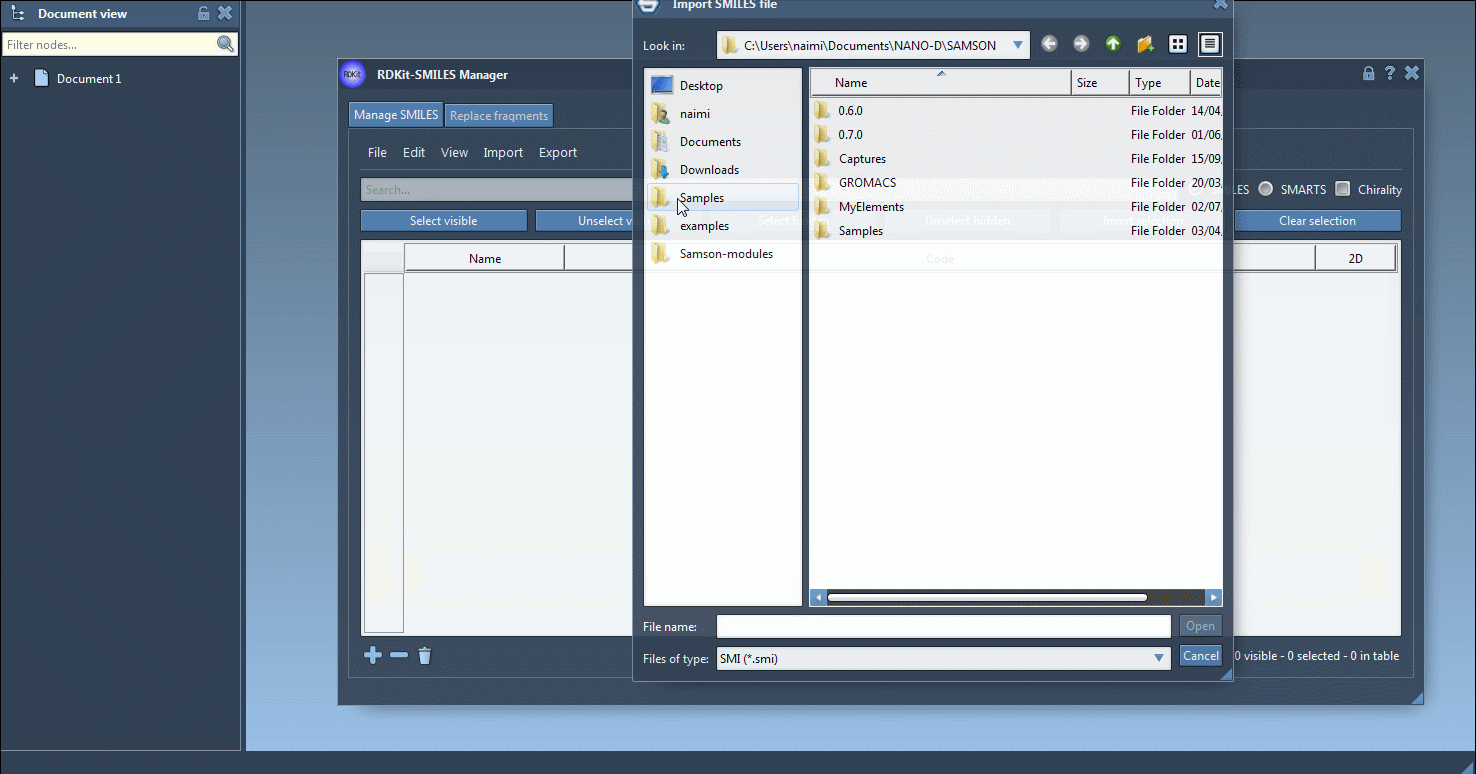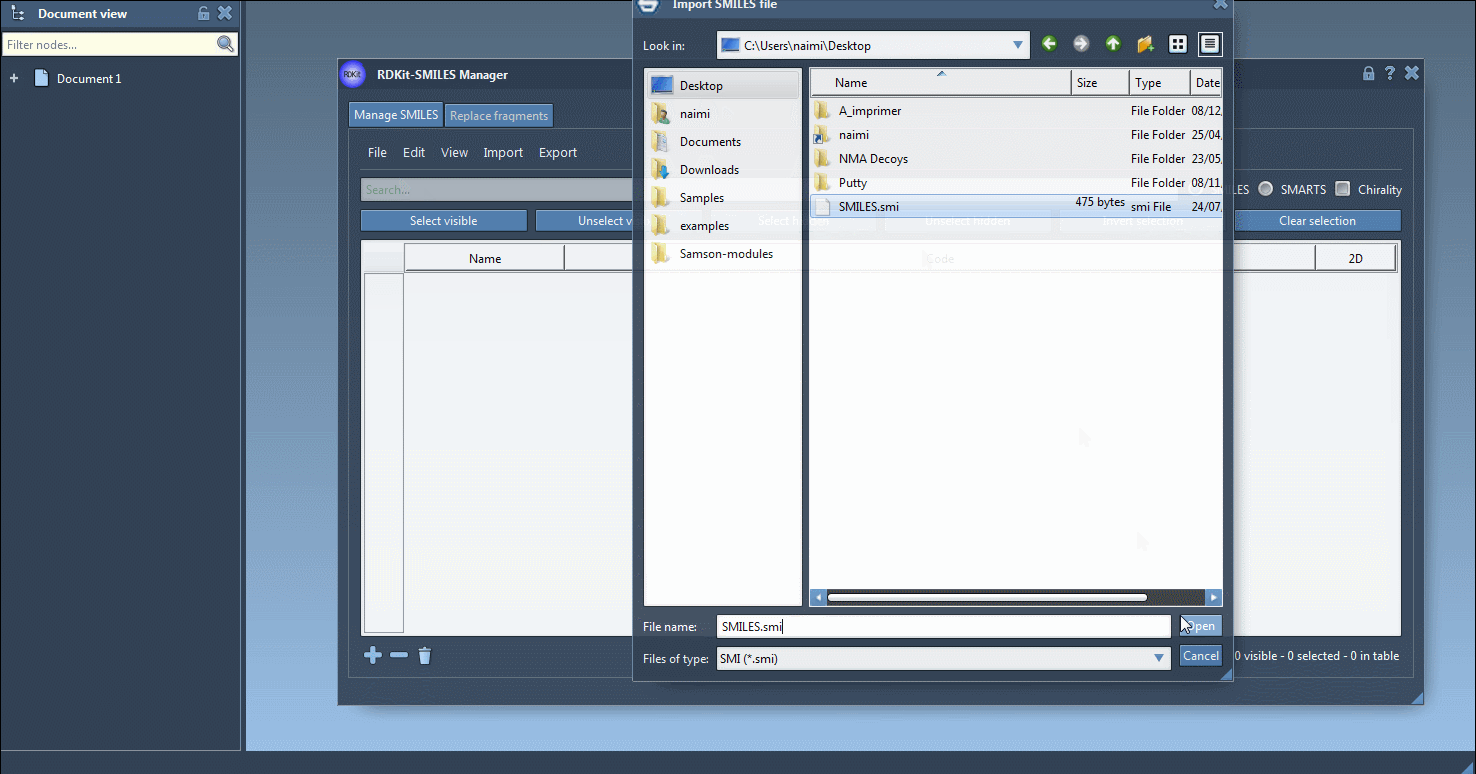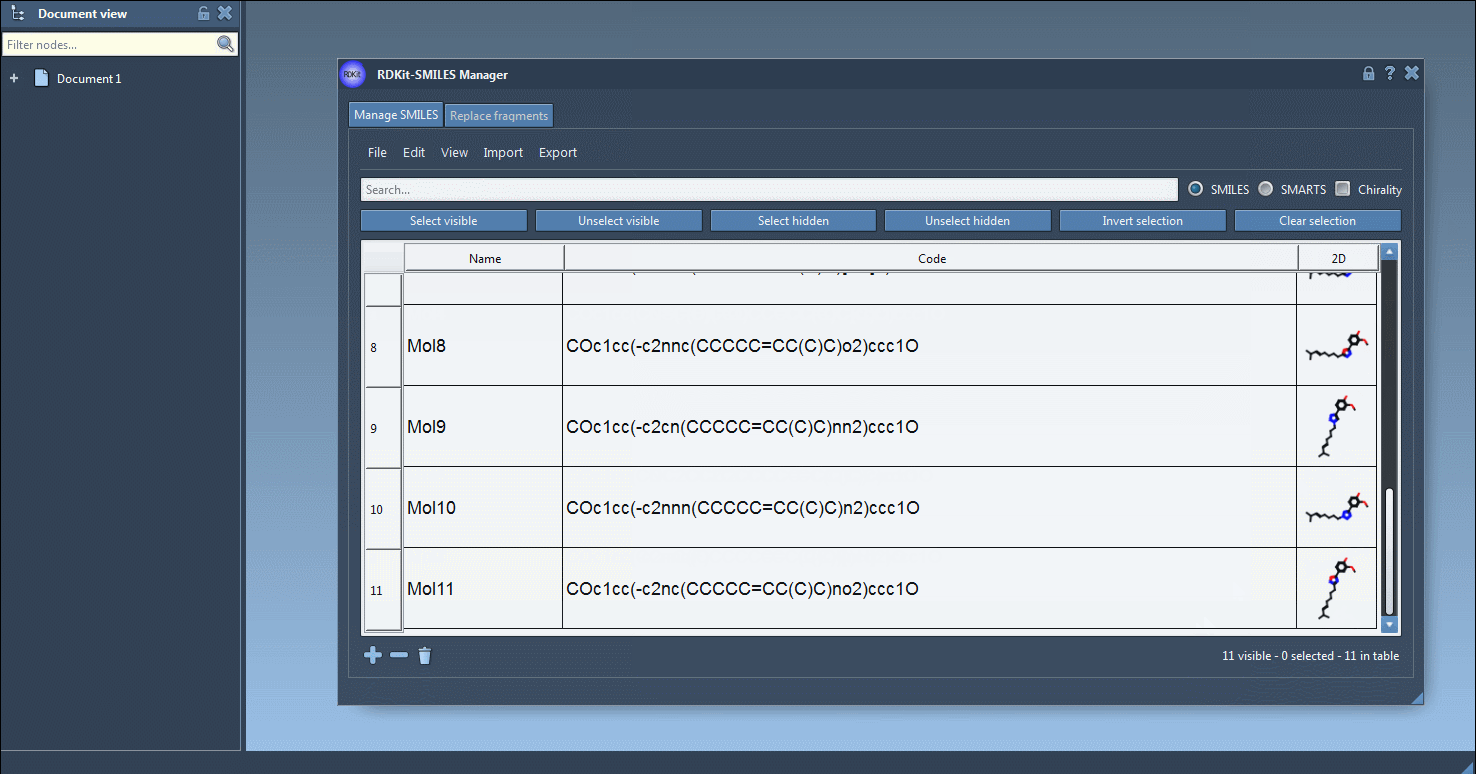
You can also add or edit SMILES strings manually, directly in the table. A molecule name can be given or automatically inferred from the SMILES code.
From SMILES to 2D and 3D structures
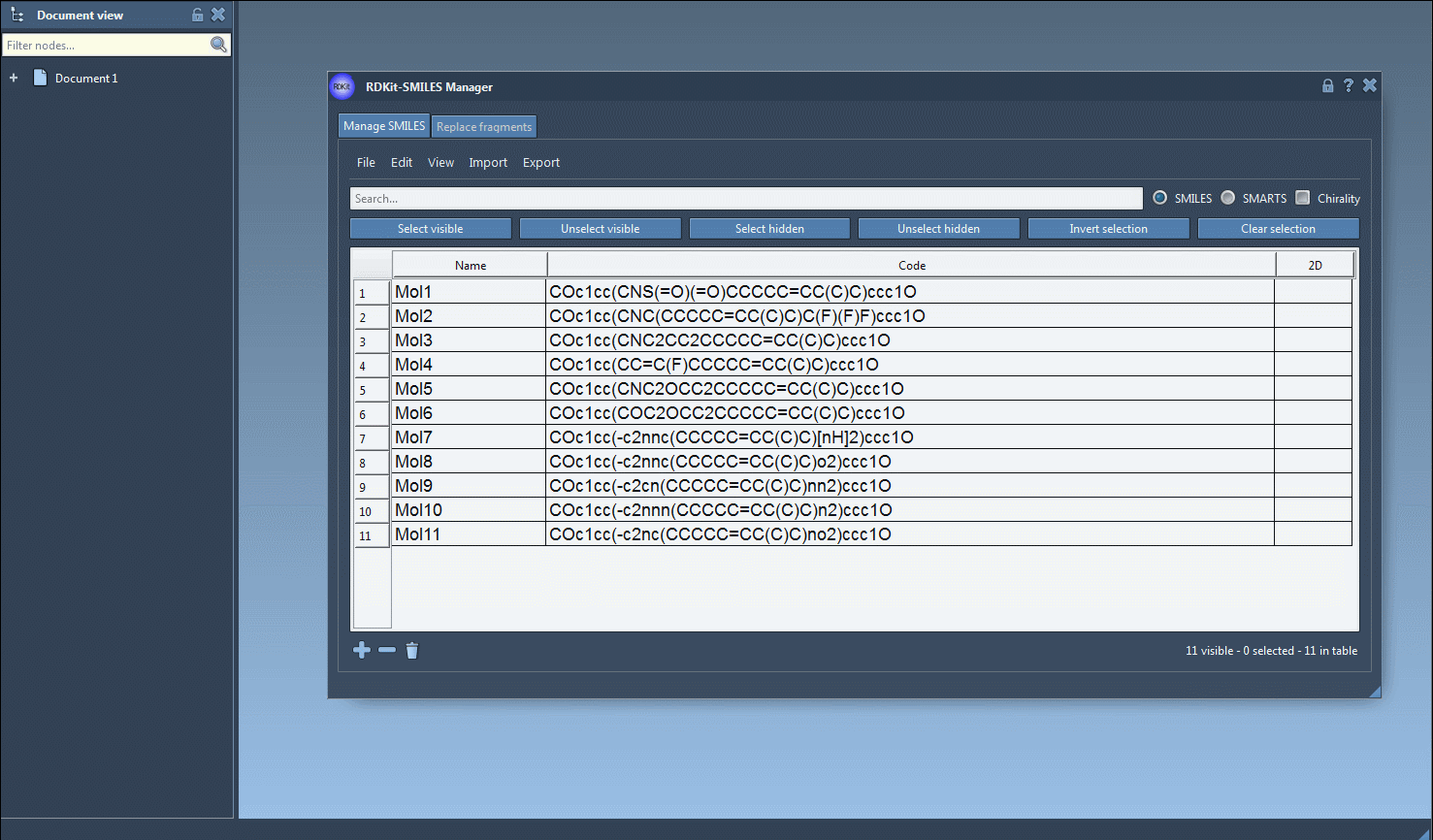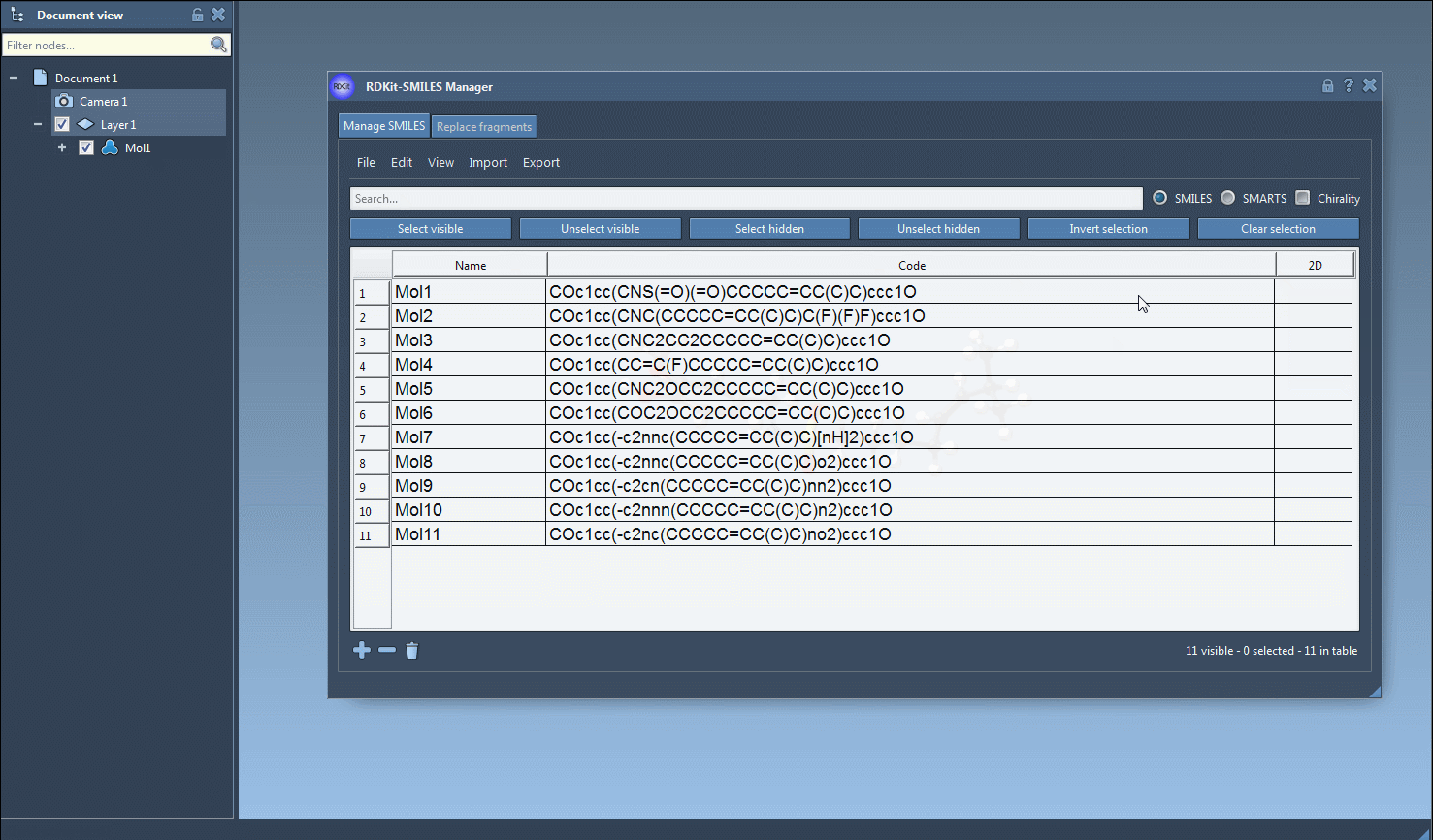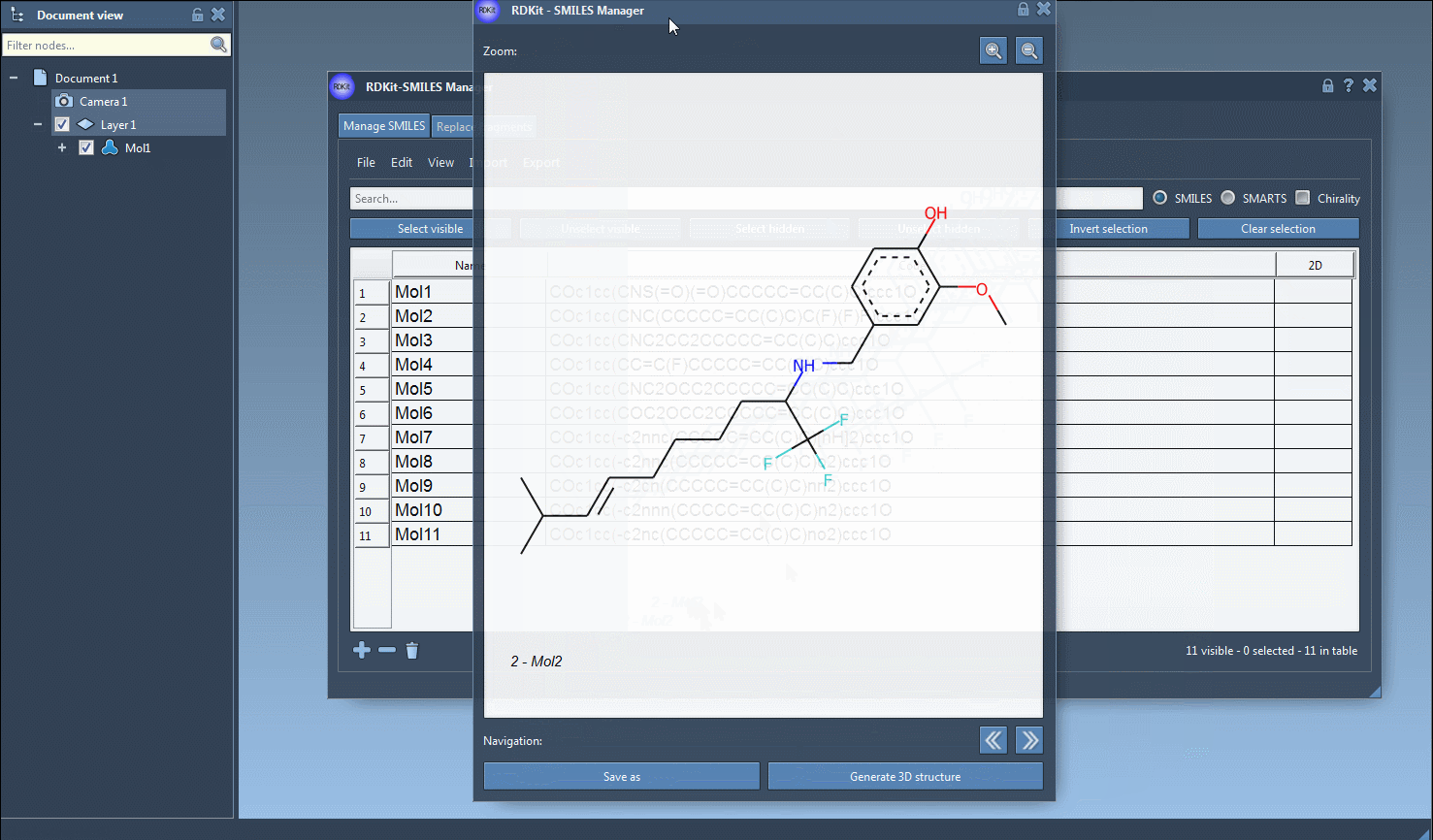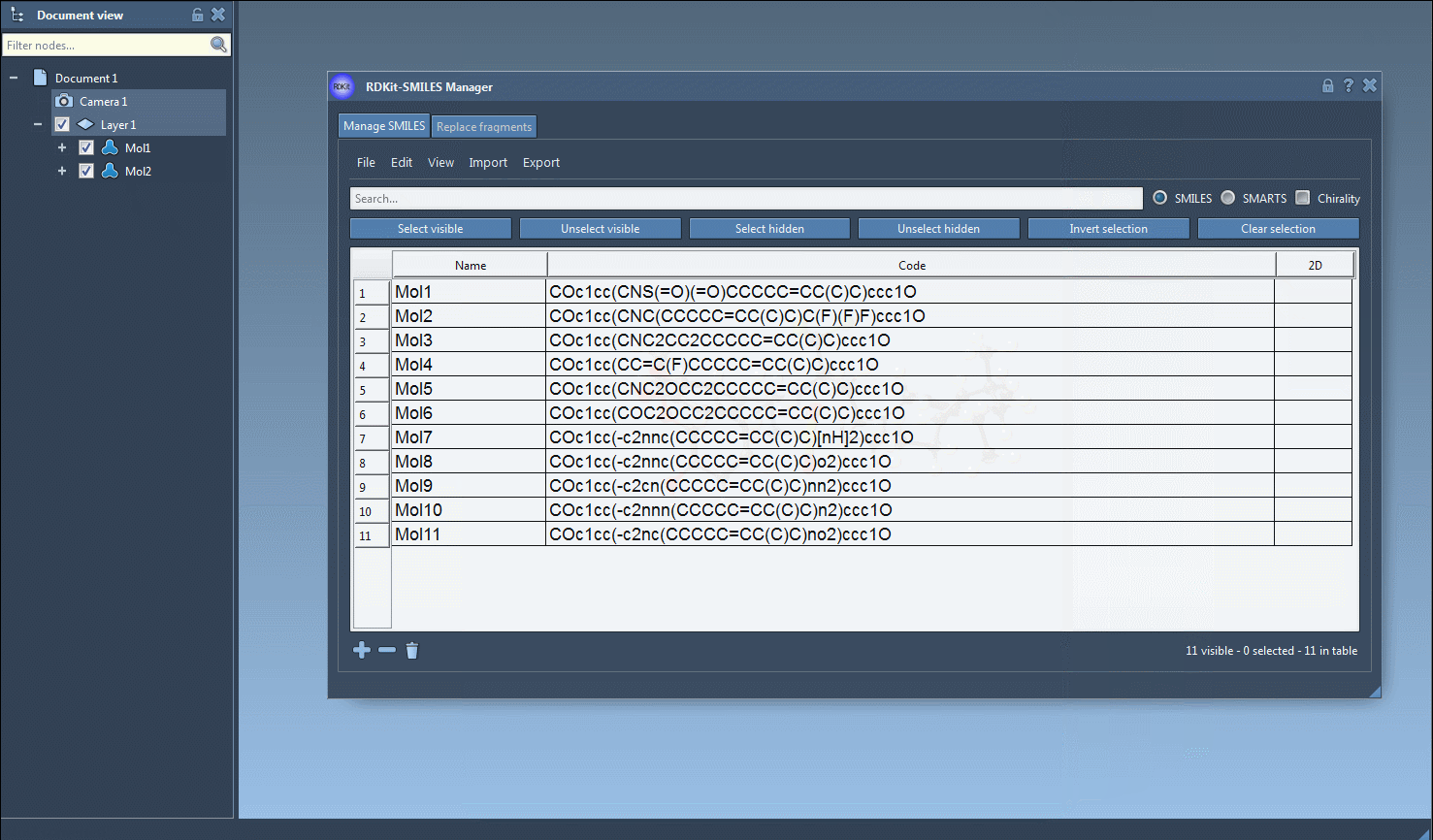
Once SMILES strings are in the table, SAMSON generates 2D depictions immediately. Invalid entries are flagged, with helpful visual cues for quick correction. Double-clicking a 2D depiction allows a larger view and optional export as PNG or SVG.

The core utility comes when converting to 3D. Select one or multiple entries, then go to Export > Selected SMILES string to Document. The 3D structures will be added to your active SAMSON document, ready to be manipulated, visualized, or further processed.
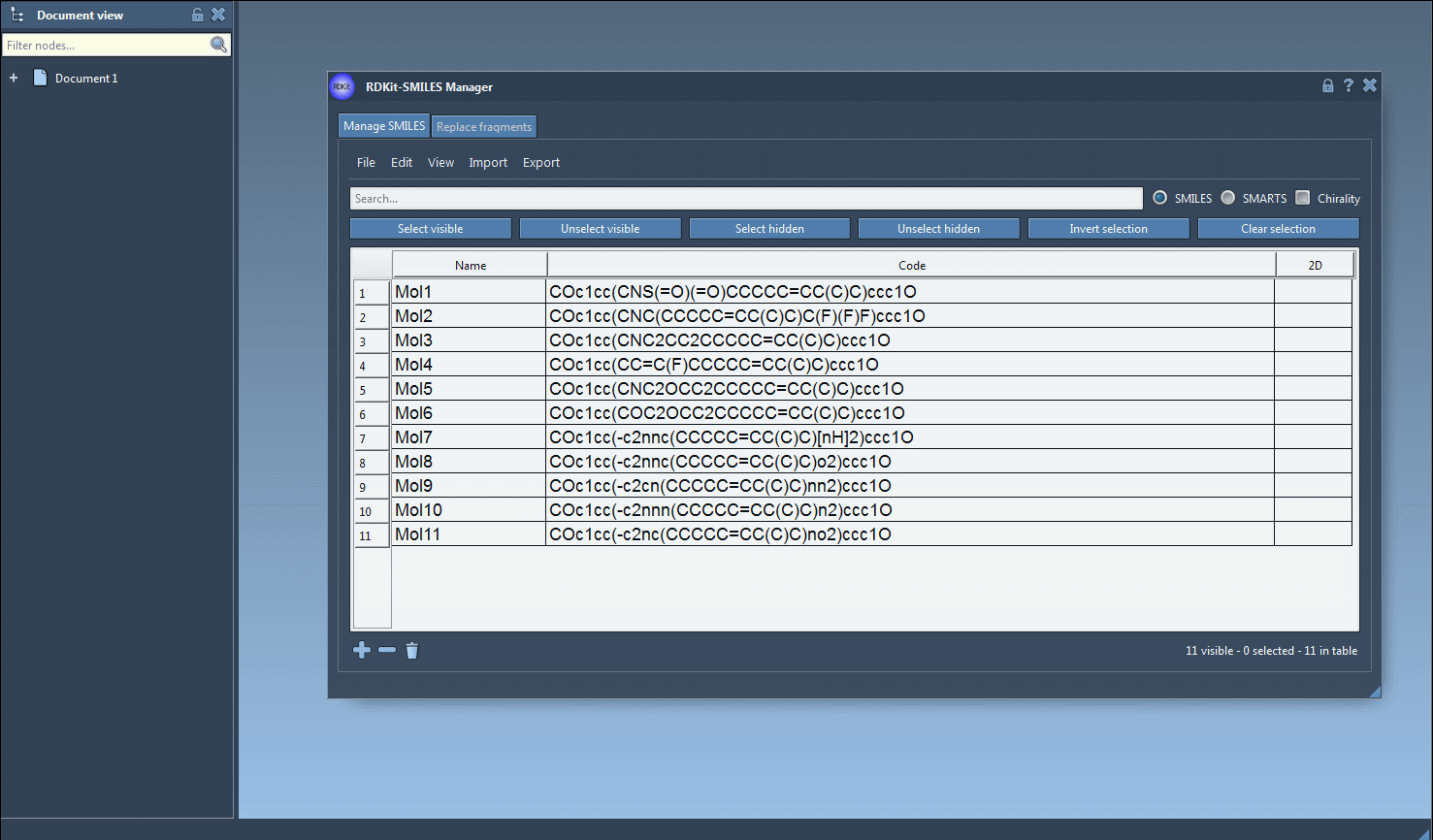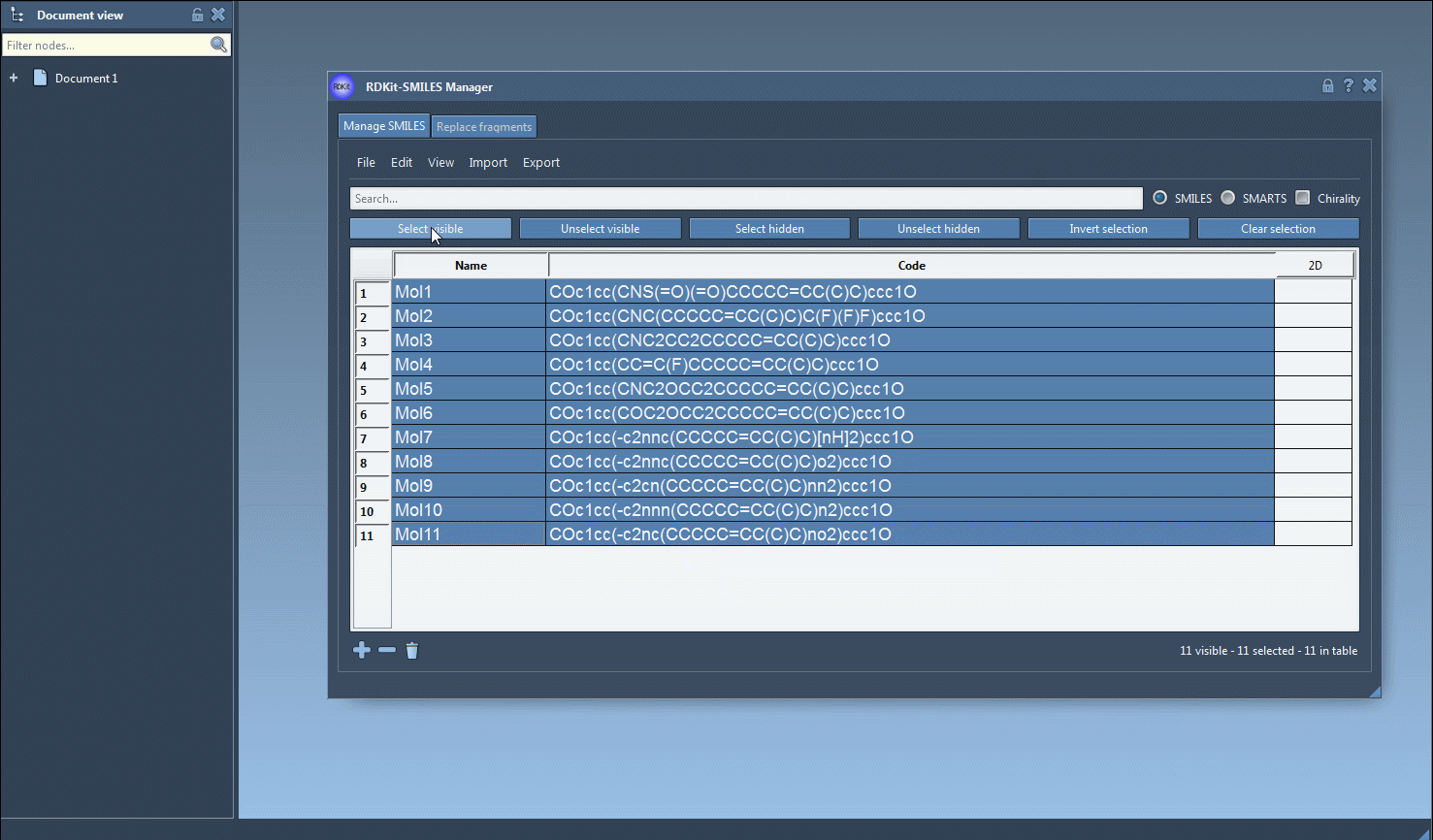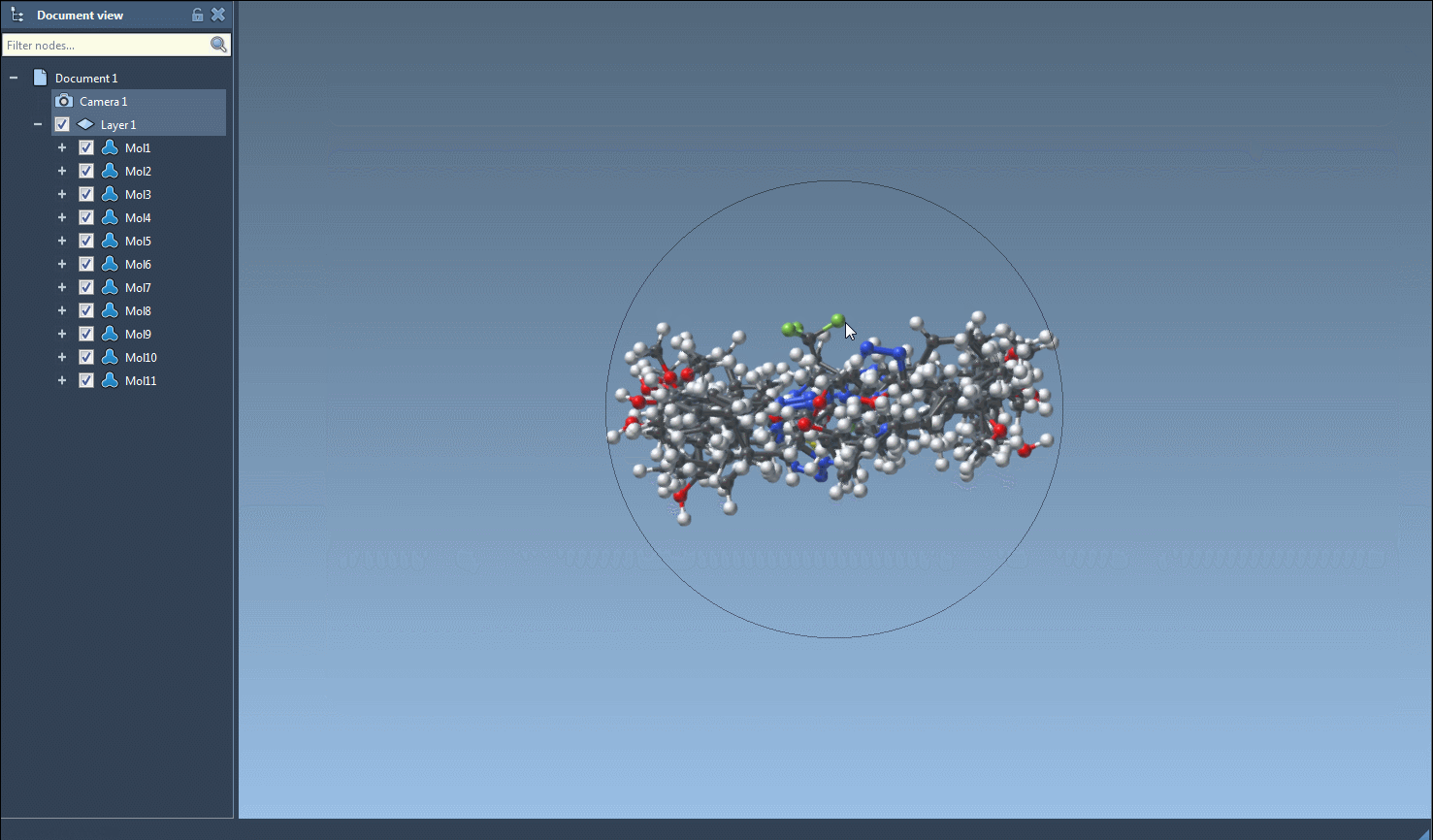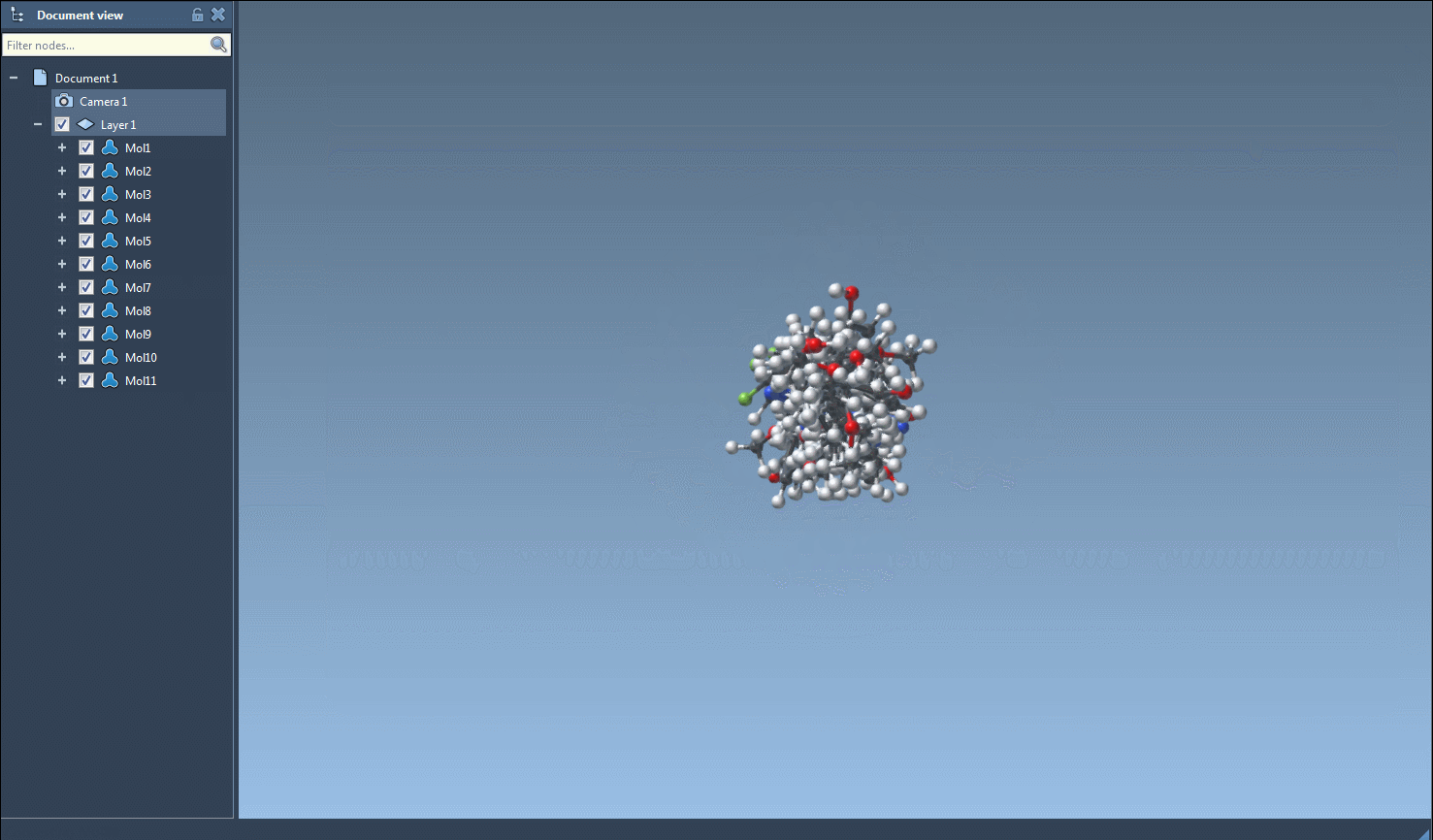
3D generation is also accessible from the right-click context menu or individual molecule view, which is convenient when validating or exploring a particular structure in greater detail.
Why it helps
Many chemistry workflows begin with a CSV, an Excel file, or a plain text file containing SMILES data. The ability to import this straight into SAMSON without conversion steps, and quickly view—and export—3D structures, enables a smoother pipeline:
- Visual validation of compound libraries before simulations
- Rapid iteration for structure-based drug design
- Integration with other SAMSON modules for processing or analysis
This makes it a helpful tool not only for advanced users but also students and researchers working on hit-to-lead or lead optimization projects who need quick feedback from molecular structures.
You can follow the full tutorial and try out more advanced features like substructure search and fragment replacement in the official documentation:
https://documentation.samson-connect.net/tutorials/smiles-manager/using-the-rdkit-smiles-manager/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.