





When it comes to molecular modeling, every minute spent on repetitive setup tasks is a minute not spent on actual research. If you regularly work with small molecules for docking studies or analog generation, you likely face the chore of initializing the structure of your molecule before you can really get started. Luckily, SAMSON offers a shortcut.
Whether you’re beginning a new project or quickly testing a hypothesis, SAMSON’s SMILES Manager makes it easy to start from a single molecule using either SMILES input or by selecting directly in the SAMSON interface.
Why it matters
Accurate molecule initialization is the first step in many cheminformatics workflows: docking, molecular dynamics, analog design, and more. But if you’re reading or copying SMILES codes from papers or internal databases, manually converting them into 2D/3D structures can be tedious, especially if you have many of them. Worse, selection errors or format issues can introduce mistakes downstream.
Two ways to get started
In the SMILES Manager interface, you’ve got two straightforward ways to define your starting molecule:
- Enter SMILES code directly: Paste in the SMILES representation of your molecule. This is ideal when copying from literature or databases.
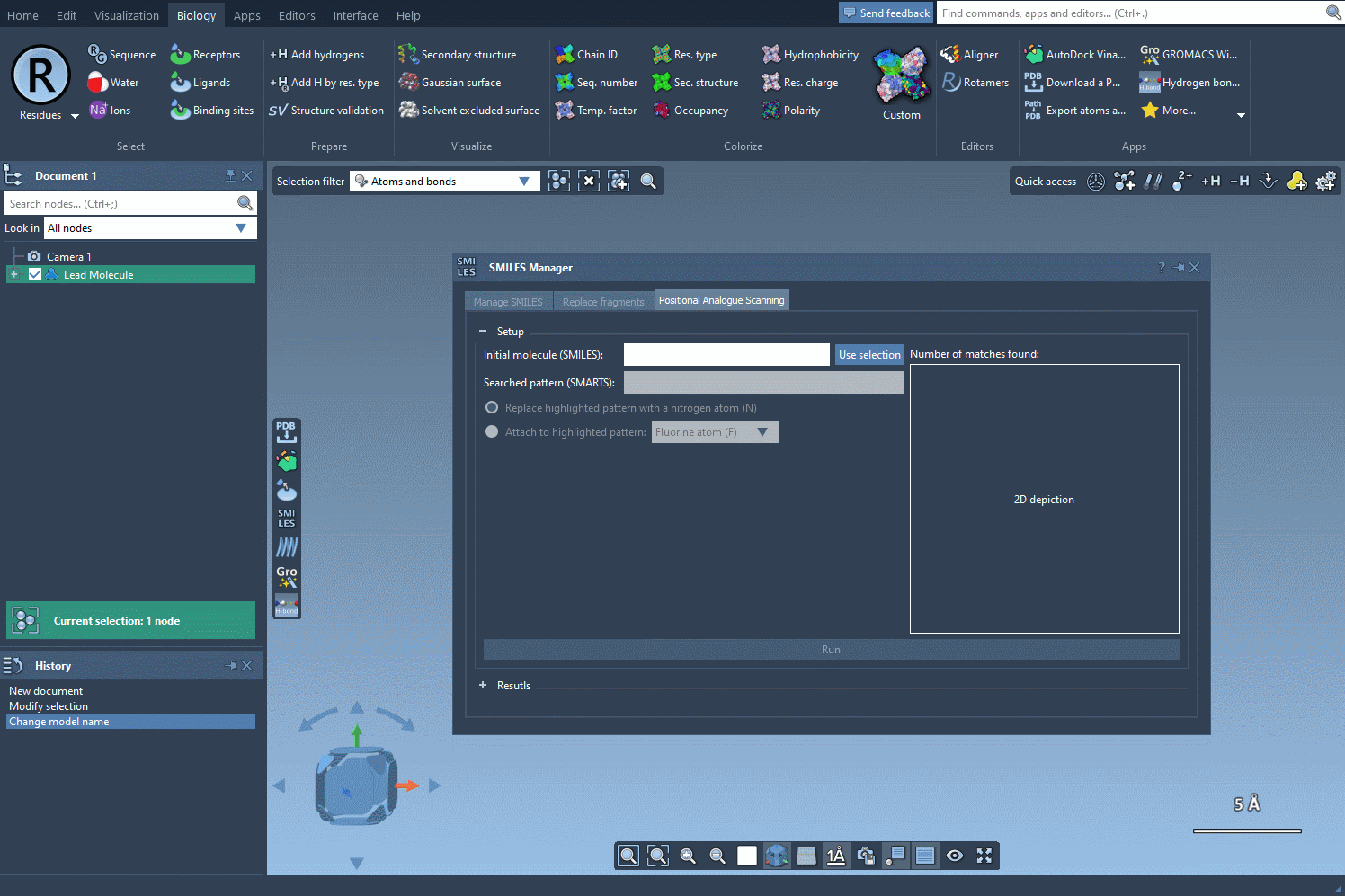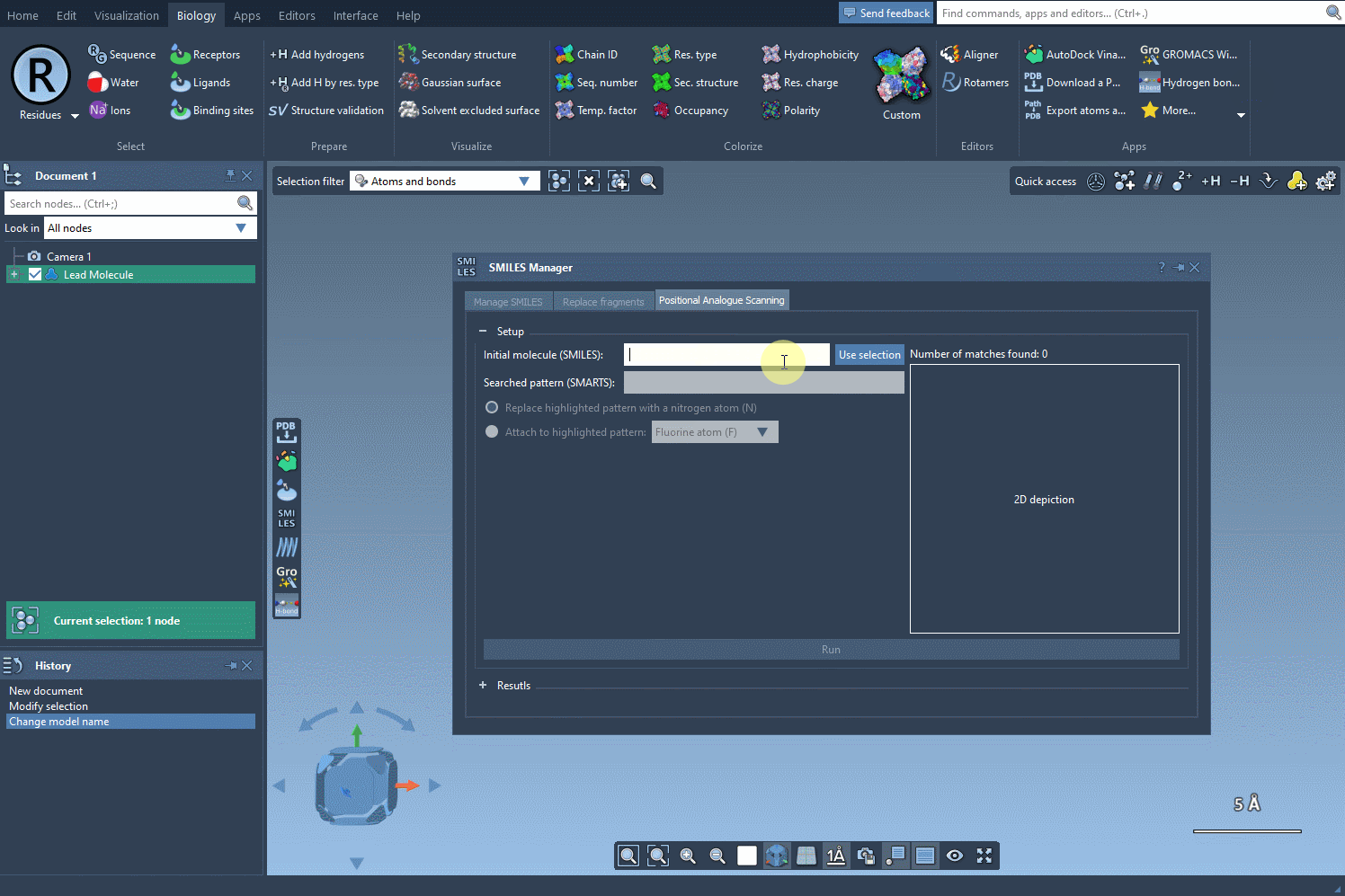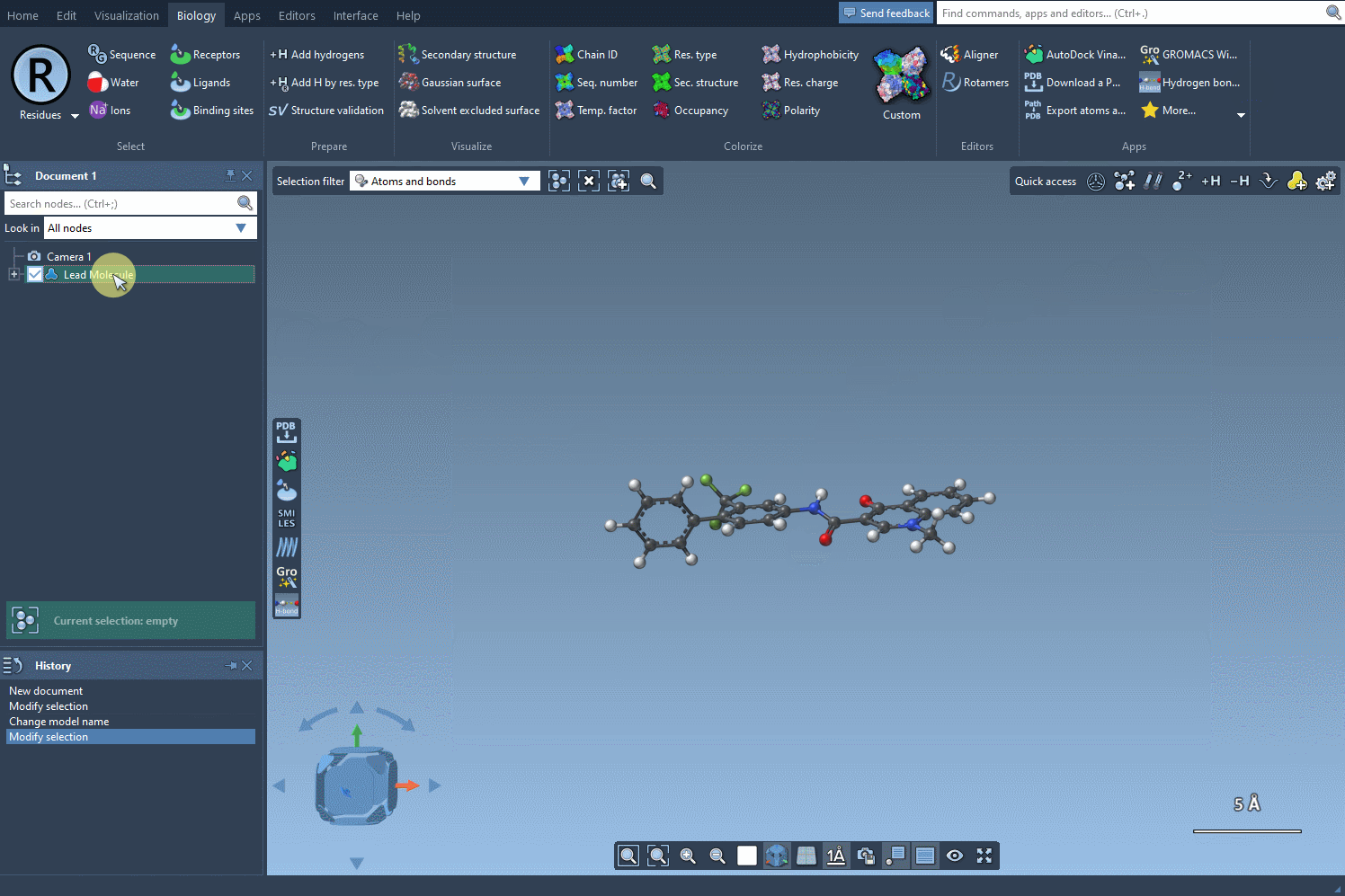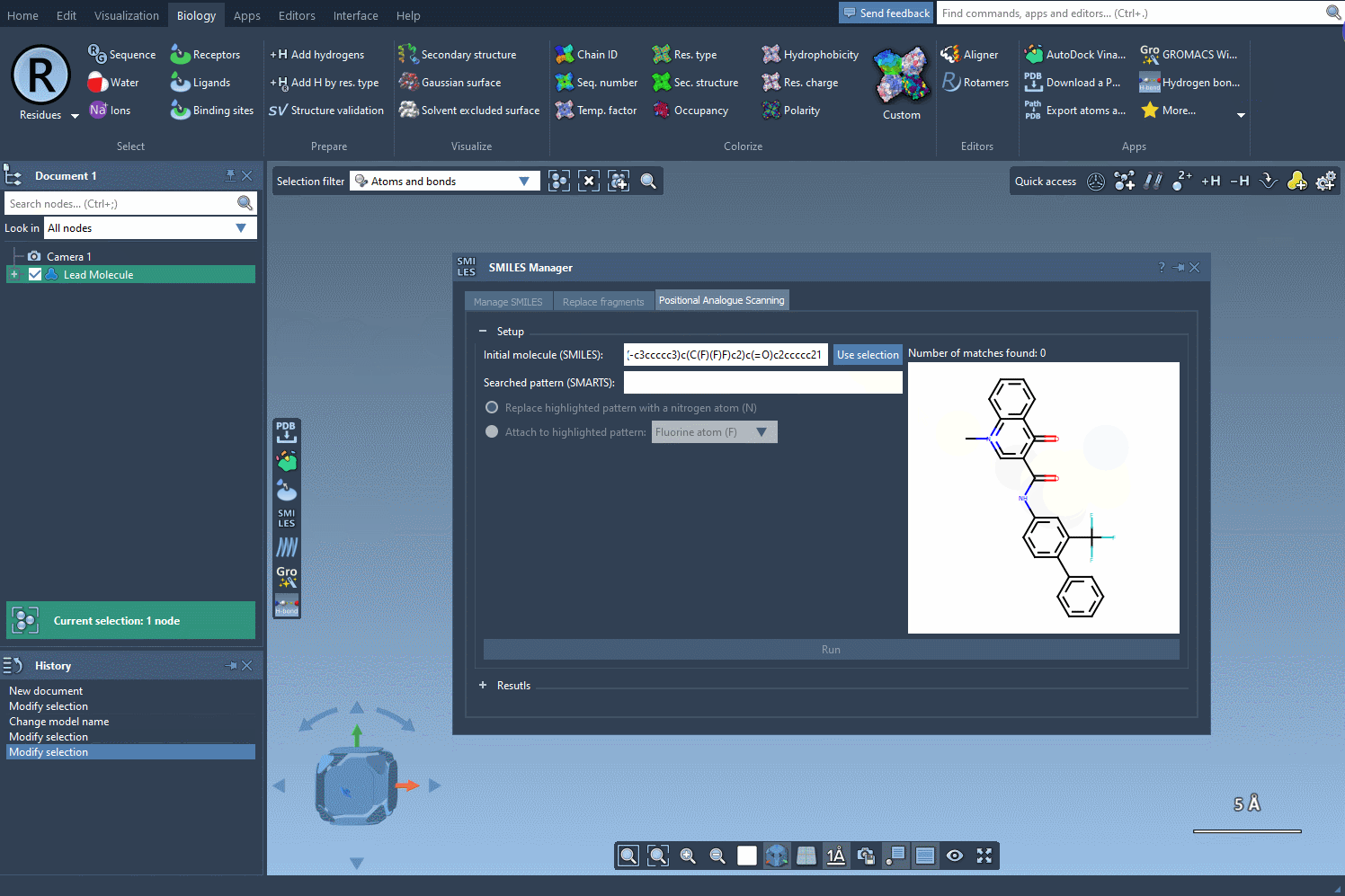
- Use selection from the SAMSON document: If you already have a molecule loaded in SAMSON (for example, from a docking or crystal structure), you can just select it and hit Use selection. No copy-pasting involved.
This small but powerful feature removes the need to switch back and forth between viewers, editors, or external conversion tools. It’s especially helpful when you’re moving quickly between experiments or loading molecules from multiple sources.
See it in action
The following animation from the documentation shows how seamless the process is:
Tip for teams and educational use
If you’re teaching molecular design or collaborating on a team project, streamlining how you input molecules ensures everyone starts from the same place. No more discrepancies due to how a structure was initialized. By encouraging standardized entry from SMILES or validated selections, you reduce setup variability and future confusion.
After entering your molecule, you’re ready to explore positional analogue scanning, docking, or structure generation. It’s a small step—but it sets the tone for more reliable and reproducible modeling sessions.
Learn more
You can find the full tutorial, including analog generation and advanced features, in the official SAMSON documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.