





Setting up a molecular simulation can sometimes feel like a road paved with pitfalls—especially when it comes to selecting and initializing a force field. For many researchers, the Universal Force Field (UFF) offers the convenience of broad coverage and applicability, but configuring it properly in a modeling environment can still be confusing. If you’re using SAMSON, this post will walk you through how to efficiently set up the Universal Force Field with minimal missteps and maximum clarity. Whether you’re simulating organic molecules, metals, or mixed systems, this guide might save you quite a bit of trial and error 💡.
Why focus on setup?
Correctly initializing UFF is a common stumbling block. Users may load a molecule and start a simulation, only to find unexpected bond orders, atom types, or even simulation crashes. This usually happens due to incomplete or incorrect perception of the molecular structure by the software. Thankfully, SAMSON includes a thoughtful set of tools to take care of these steps during setup—if you know where to look and what to adjust.
How to properly set up UFF in SAMSON
Once you’ve opened a document with your molecular system, follow this path to add UFF:
- Go to Edit > Simulate > Add simulator or use the shortcut Ctrl+Shift+M (on macOS: Cmd+Shift+M).
- Choose Universal Force Field from the interaction model list.
- Select a state updater like FIRE to control geometry relaxation.
- Click OK to advance.
This will bring up a setup window with crucial options. One critical choice is:
- Use existing bonds: Selecting this checkbox means you’re relying on already-defined bonds. If unchecked, SAMSON will recalculate bonds based on atom types and positions.
After clicking OK, SAMSON executes the following perception steps:
- Bonds are determined (if not already present).
- Bond orders are computed.
- Atom types are assigned.
Should there be inconsistencies—such as ambiguous valences or unknown atom types—a warning will appear. Taking the time to verify bond distances and molecular geometry beforehand can help prevent these alerts.
Visual feedback during setup
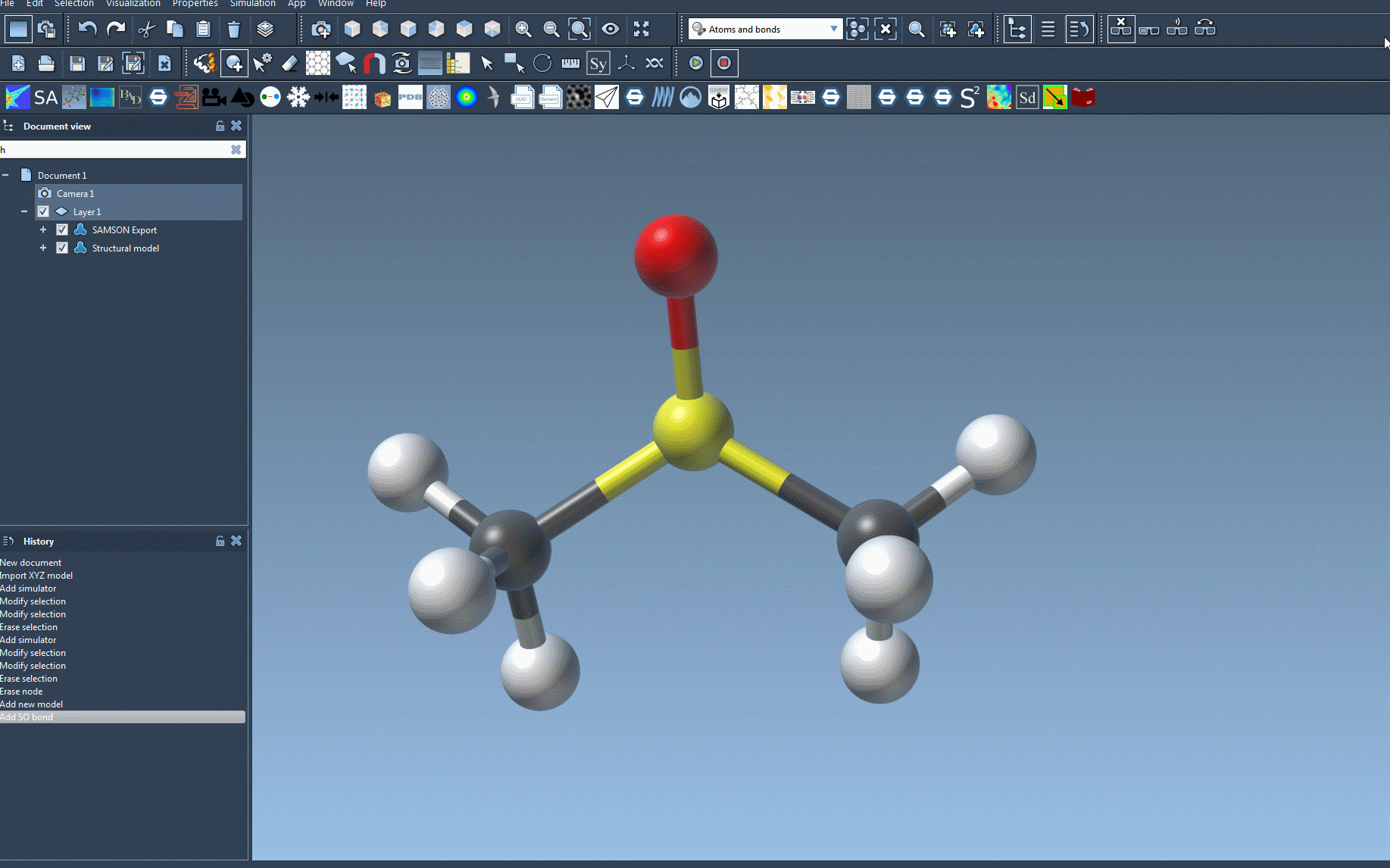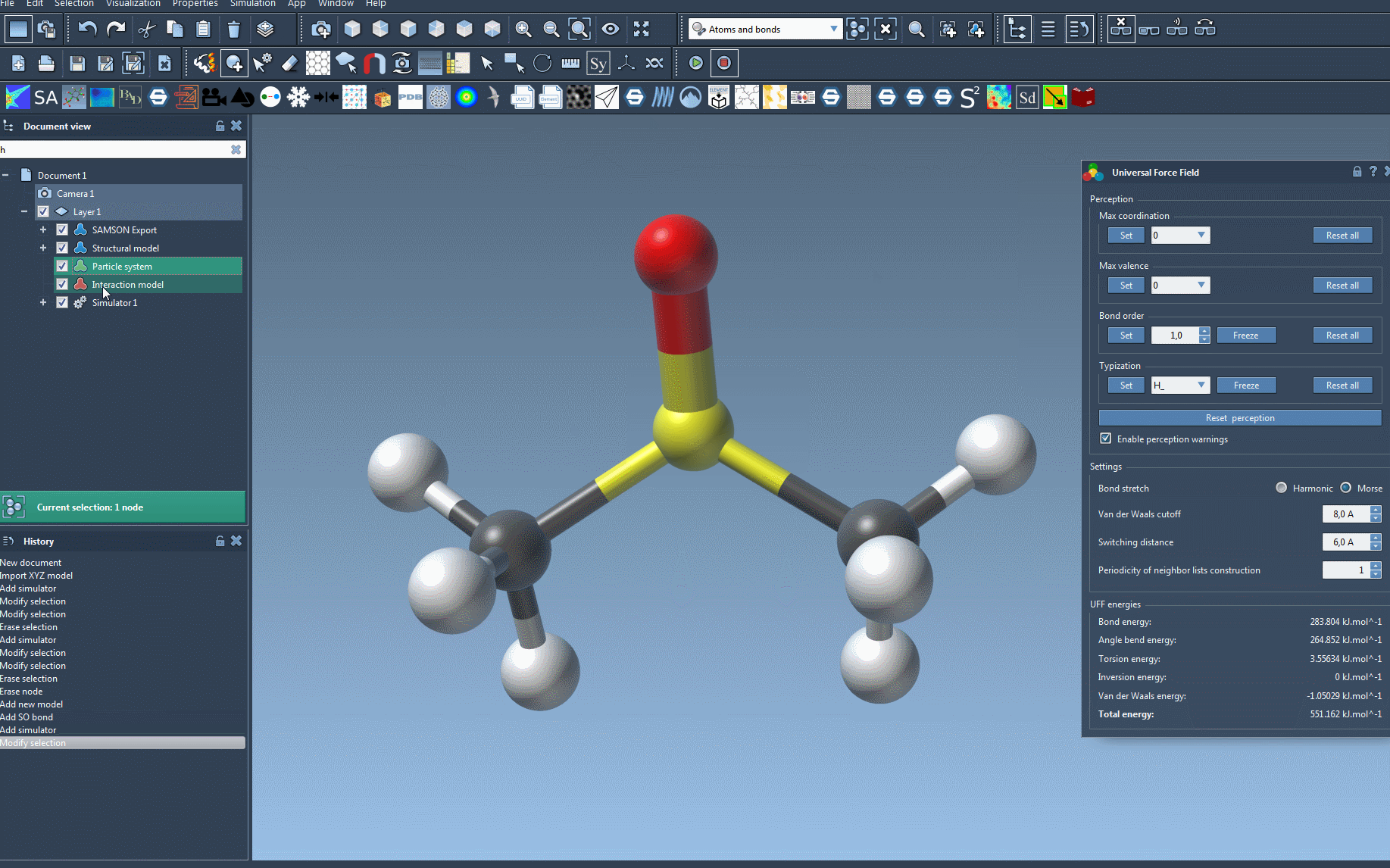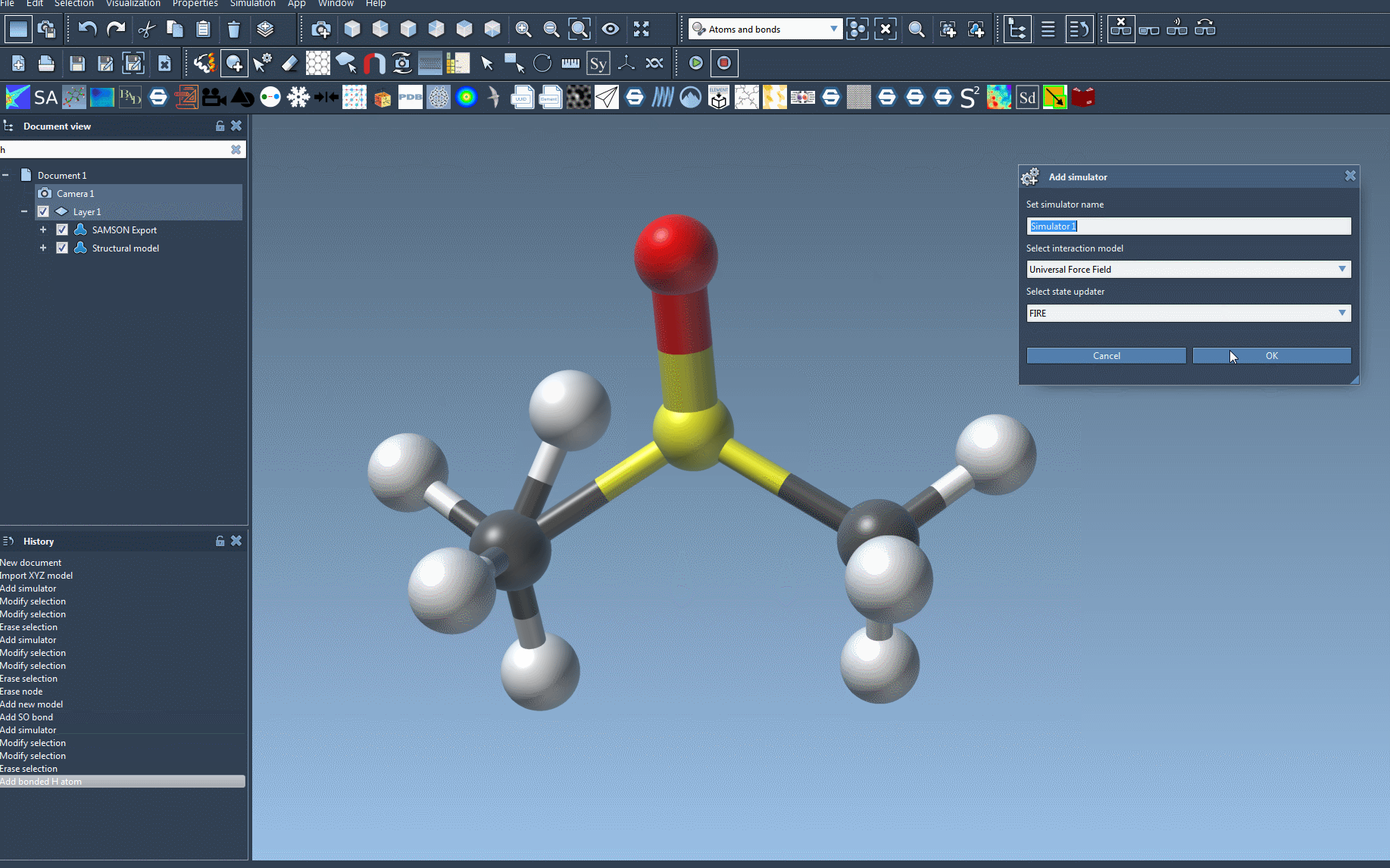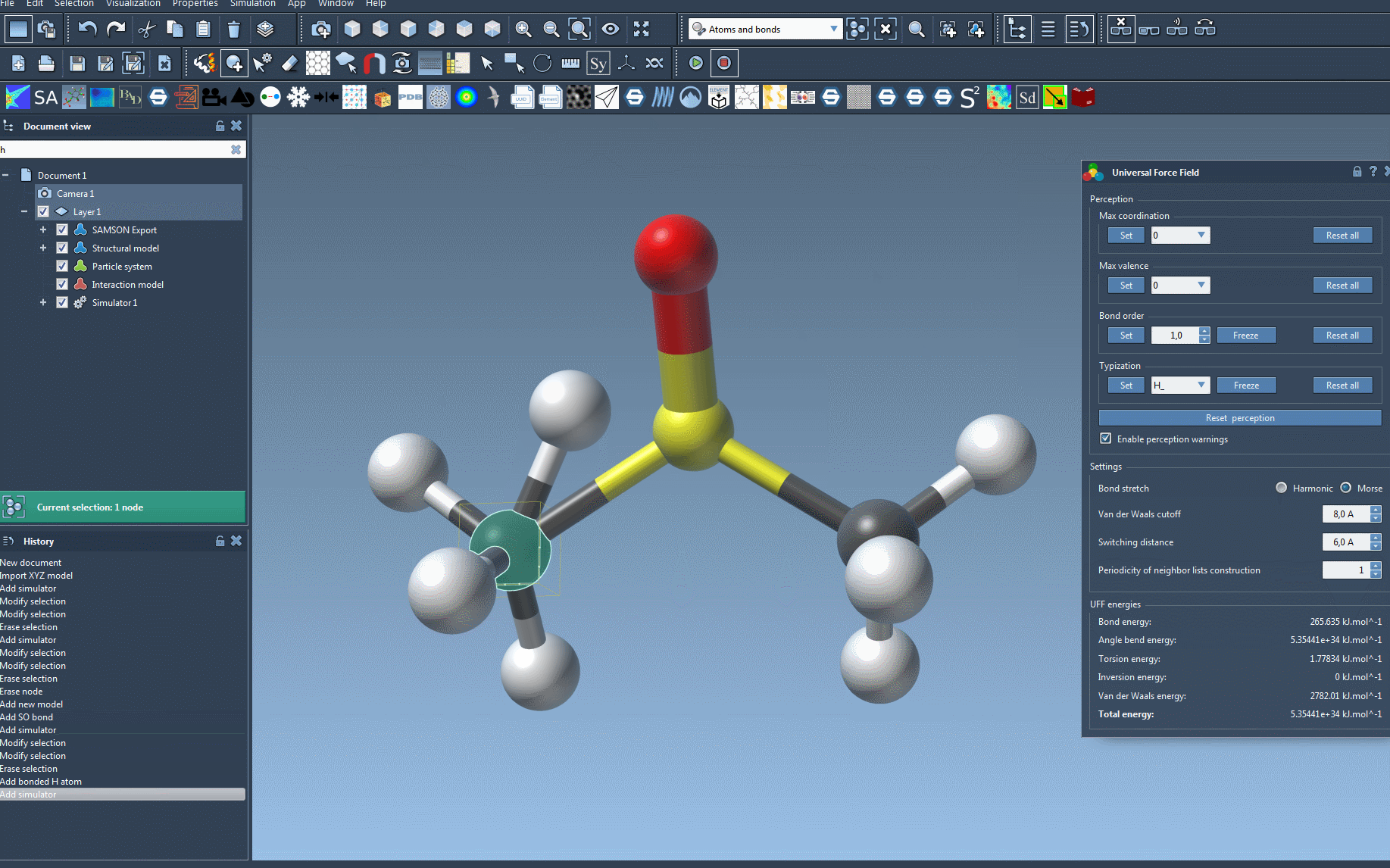
After initializing, a parameter window appears where you can control various settings, including:
- Harmonic vs. Morse bond stretch models
- Van der Waals cutoff and switching distances
- Neighbor list update frequency
This window also displays energy contributions from different force field terms, which can help diagnose issues or confirm the system is behaving as expected.
Tip: A clean setup paves the way for better simulations
Especially for larger or more complex systems, building the model carefully and choosing the right options during UFF setup can help you avoid downstream issues during simulation or optimization. Rechecking bond lengths, avoiding overlapping atoms, and understanding what perception means in this context will make your simulations more reliable.
Learn more and explore detailed options in the full documentation page: https://documentation.samson-connect.net/tutorials/uff/uff/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.