





Defects play a major role in the properties of crystalline materials: they can influence conductivity, hardness, optical behavior, and more. Yet, modeling them can be time-consuming if you’re not equipped with the right tools. If you’re exploring how imperfections affect the structure of diamond or similar crystals, this guide introduces a practical and visual way to simulate defects with the SAMSON Crystal Creator app.
Why simulate crystal defects?
Perfect crystals are a theoretical ideal. In reality, materials often contain vacancies, substitutions, and other types of disorder, which strongly influence their mechanical, electronic, and chemical properties. Being able to model such imperfections helps in designing materials with specific characteristics, whether for semiconductors, alloys, or nanostructures.
Let’s take diamond, renowned for its structured lattice of carbon atoms. Want to introduce atomic defects and observe the changes? Here’s how to do it in SAMSON.
Step-by-step: modeling defects in diamond
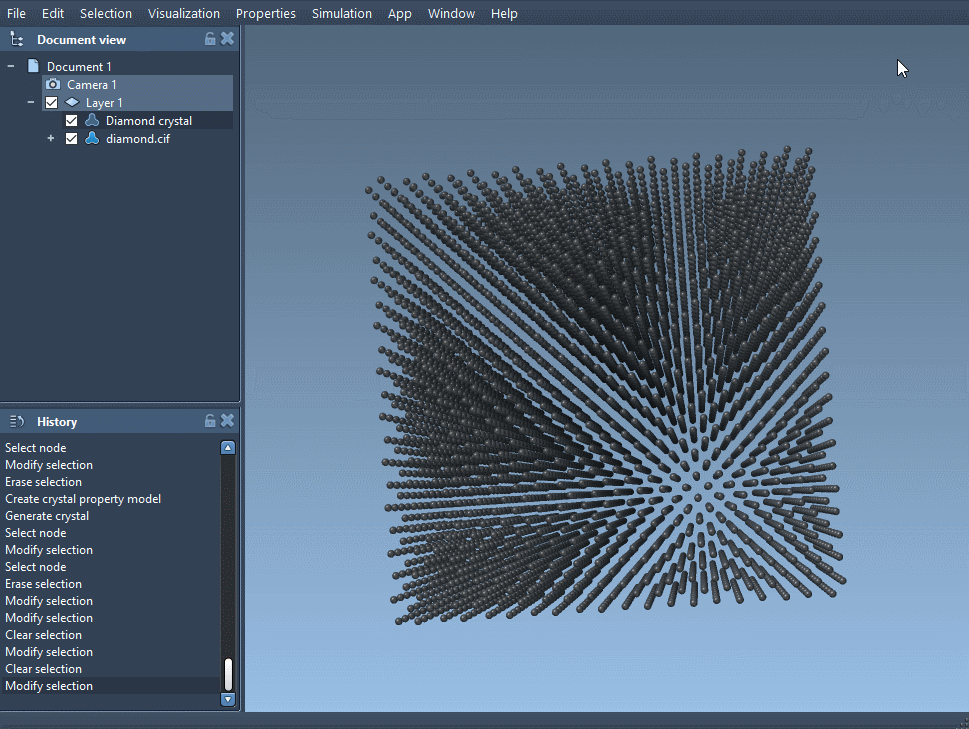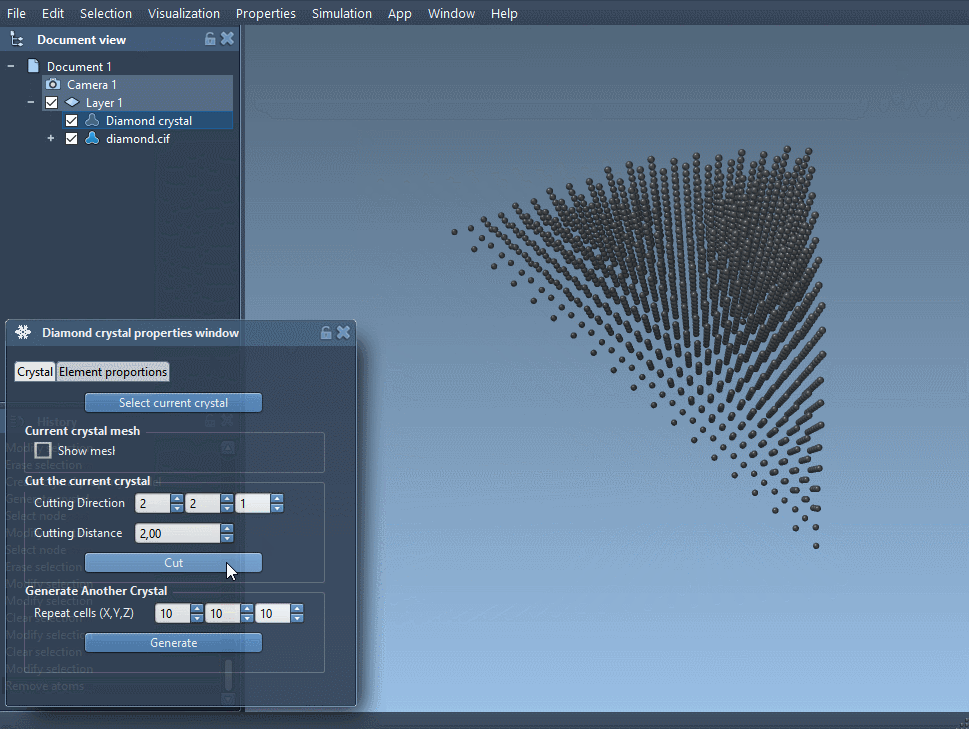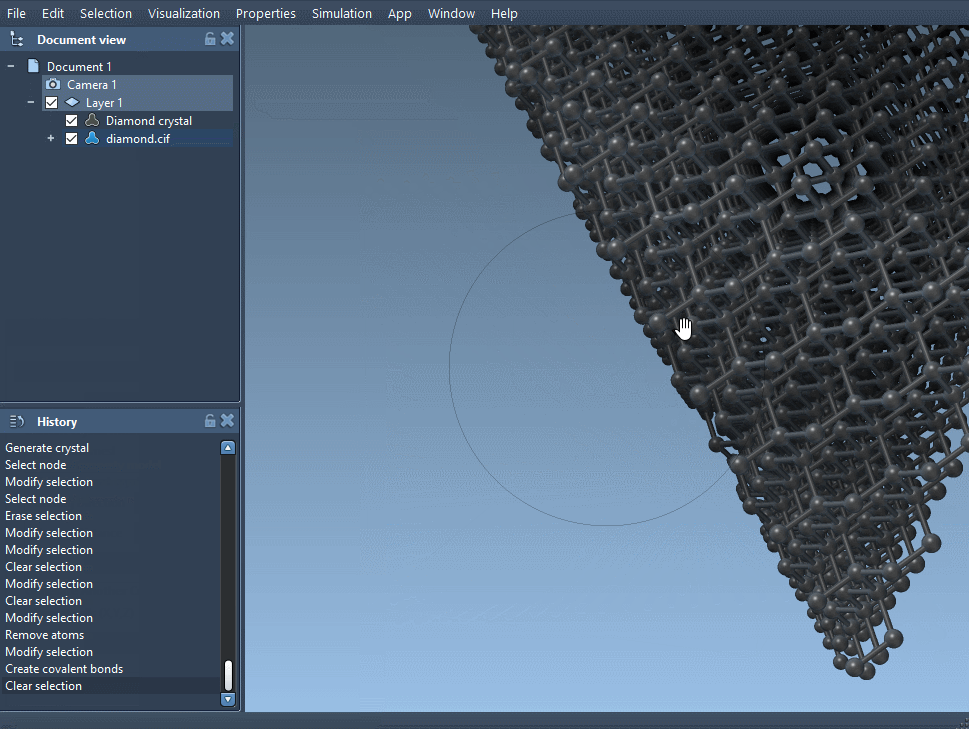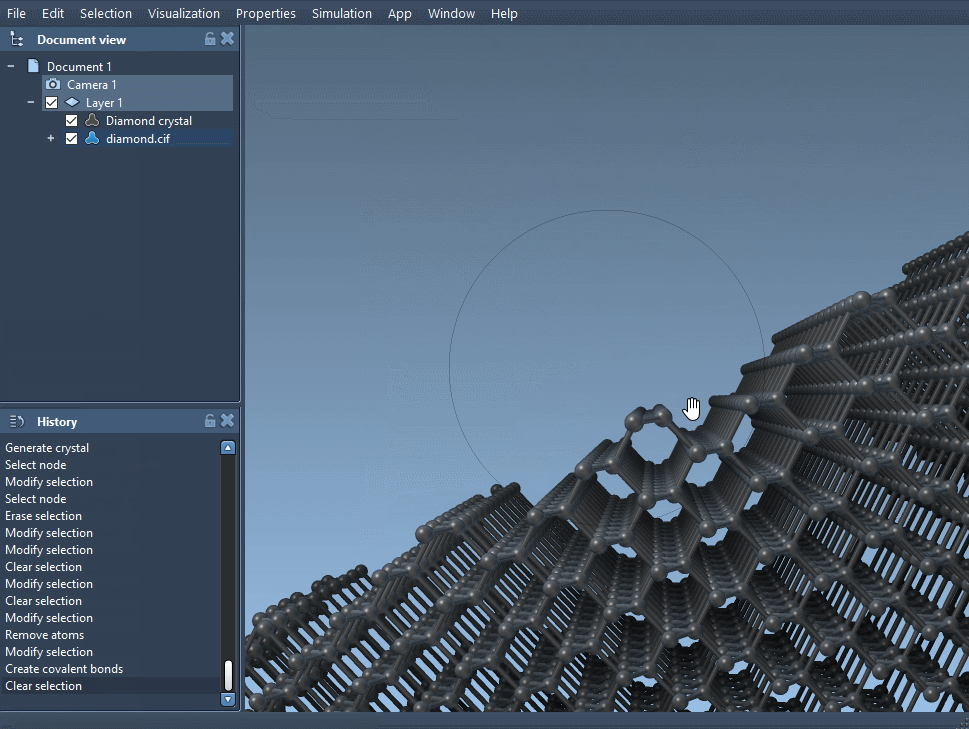
- Load the diamond CIF file
You can download a diamond structure from a CIF database like RRUFF, and load it into SAMSON using the Crystal Creator app. If you need help importing CIF files, check the User guide. - Create bonds
After opening the file, create the bonds to visualize the full structure. Without this, the atoms may appear as unconnected spheres. - Minimize the structure (optional)
Use the Brenner interaction model to perform an energy minimization step, allowing atoms to relax into a stable configuration. This is especially helpful before and after introducing defects, so you can visually assess how the structure changes. - Edit the CIF to introduce defects
Make a copy of your CIF file and open it in a text editor. Locate the section that lists the atom positions, typically structured in this way: - Add occupancy values
To simulate a partial vacancy, adjust the format to include occupancy values. For instance: - Reload in SAMSON
Back in SAMSON, open the modified CIF file and, again, create the bonds. You should notice subtle (or not-so-subtle) differences in the atomic arrangement based on the probabilities you’ve set. You can now analyze how missing atoms affect bond lengths, coordination, or local symmetry.
|
1 2 3 4 5 6 |
loop_ _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z C 0.00000 0.00000 0.00000 |
|
1 2 3 4 5 6 7 |
loop_ _atom_site_label _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _atom_site_occupancy C 0.00000 0.00000 0.00000 0.95 |
This line means the carbon atom has a 95% chance of being present in that site. Multiple atoms can have different occupancies to simulate complex defect patterns.
What does this offer you?
Instead of relying on complex scripts or external preprocessing tools, SAMSON provides a direct and visual pipeline to:
- Modify atomic occupancies manually,
- Apply force fields to study behavior under defects,
- Explore realistic variations in structural geometry due to imperfections.
Although demonstrated with diamond, this approach works with any crystal structure, making it a versatile tool in the hands of molecular modelers and materials scientists alike.
To learn more, visit the full guide here: https://documentation.samson-connect.net/tutorials/crystal-creator/generating-crystal-models/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.