





When exploring large-scale conformational changes in proteins, molecular modelers often face the same bottlenecks: visualizing smooth transition paths is time-consuming, and most available tools either require heavy computation or offer limited interactivity. This can slow down studies related to conformational analysis, umbrella sampling setups, or even preliminary transition-state exploration.
If you’ve been manually tweaking intermediate states or waiting for simulation results to derive transition paths, you might want to take a closer look at the ARAP Interpolator extension in SAMSON. This simple yet powerful app can generate realistic, smooth interpolation paths between two protein structures in just a few seconds — a handy way to navigate from Structure A to Structure B without manually modeling every step.
Working With Clean Conformations
To get reliable results with the ARAP Interpolation App, the two input structures should be preprocessed. Remove waters, ions, ligands, and alternate locations to ensure only the biologically relevant parts are included. In the example below, two conformations of the Diphtheria Toxin (1DDT and 1MDT) are used:
- Load both PDBs –
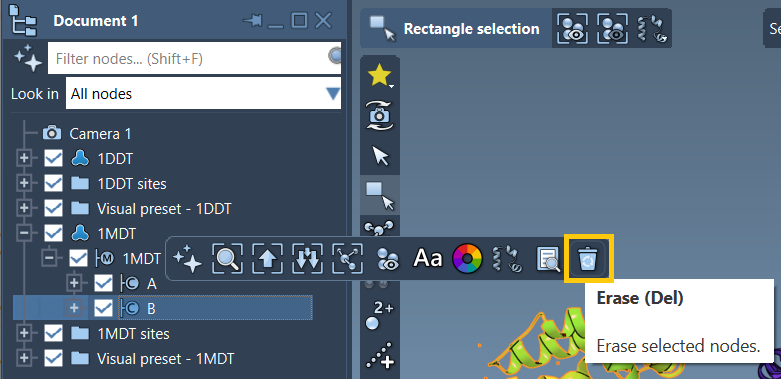
1DDTand1MDT– using the Home > Fetch panel in SAMSON. - Delete non-relevant chains (e.g., chain
B) to isolate the chain of interest (e.g., chainA). - Use Home > Prepare to clean up structures automatically.
Conformations: The Building Blocks of a Transition Path
Once the protein structures are cleaned and simplified, you can create conformations in SAMSON:
- Select the structure in the Document view
- Go to Edit > Conformation and name it based on the PDB and chain (e.g.,
1DDT A)
Repeat this for the goal structure. These conformations will represent the start and end states of the interpolated path.
Configure ARAP for Intuitive Interpolation
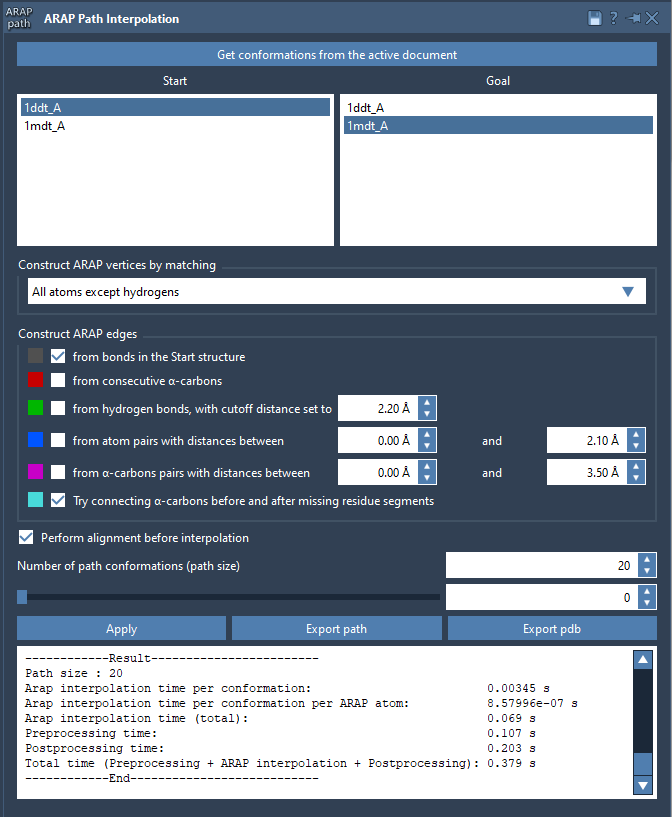
Once the conformations are ready, launch the app from Home > Apps > Biology > ARAP Path Interpolation. The interface is straightforward:
- Use Get conformations from the active document
- Select your start and goal conformations
For most proteins, it’s effective to match atoms using the option All except hydrogens and build ARAP edges from bonds in the start structure. You may also choose to connect α-carbons across missing segments to ensure continuity in regions with unresolved residues.
Finally, setting the number of path conformations to a value like 20 gives enough resolution across the transition.
From Transition to Visualization
Once the interpolation is run, use the slider to preview intermediate states along the path:
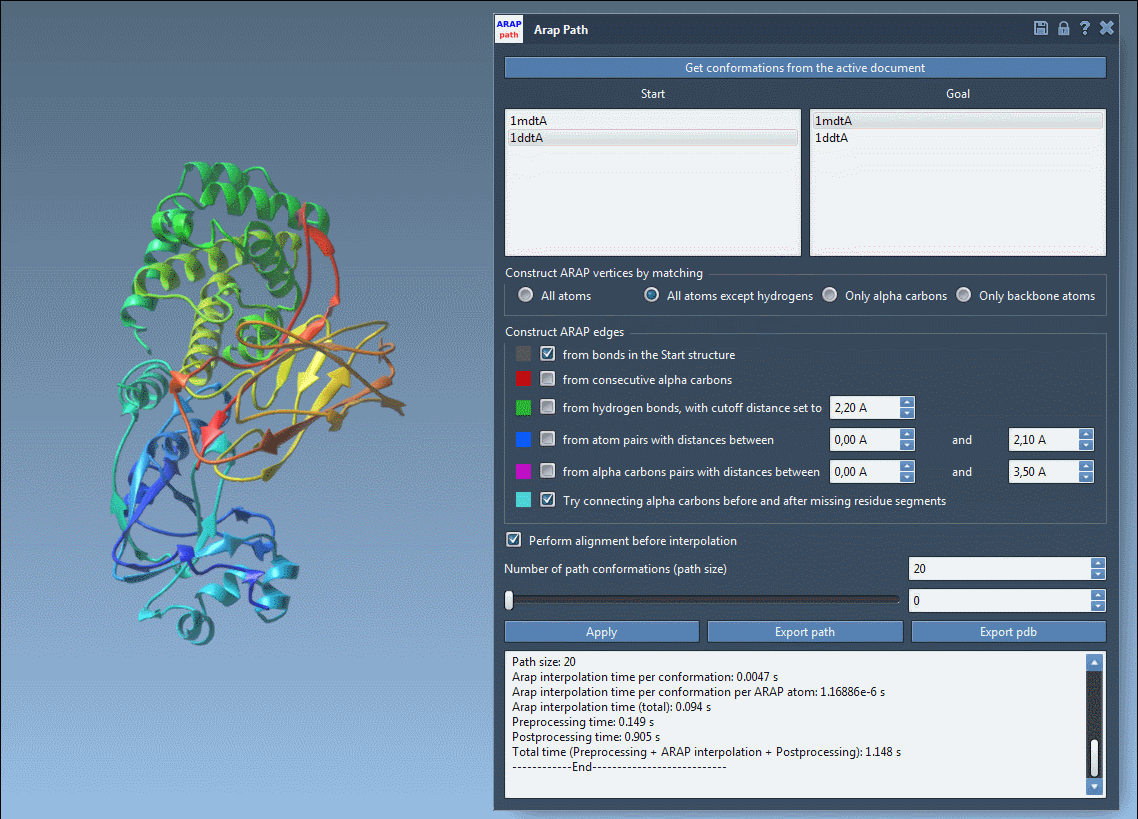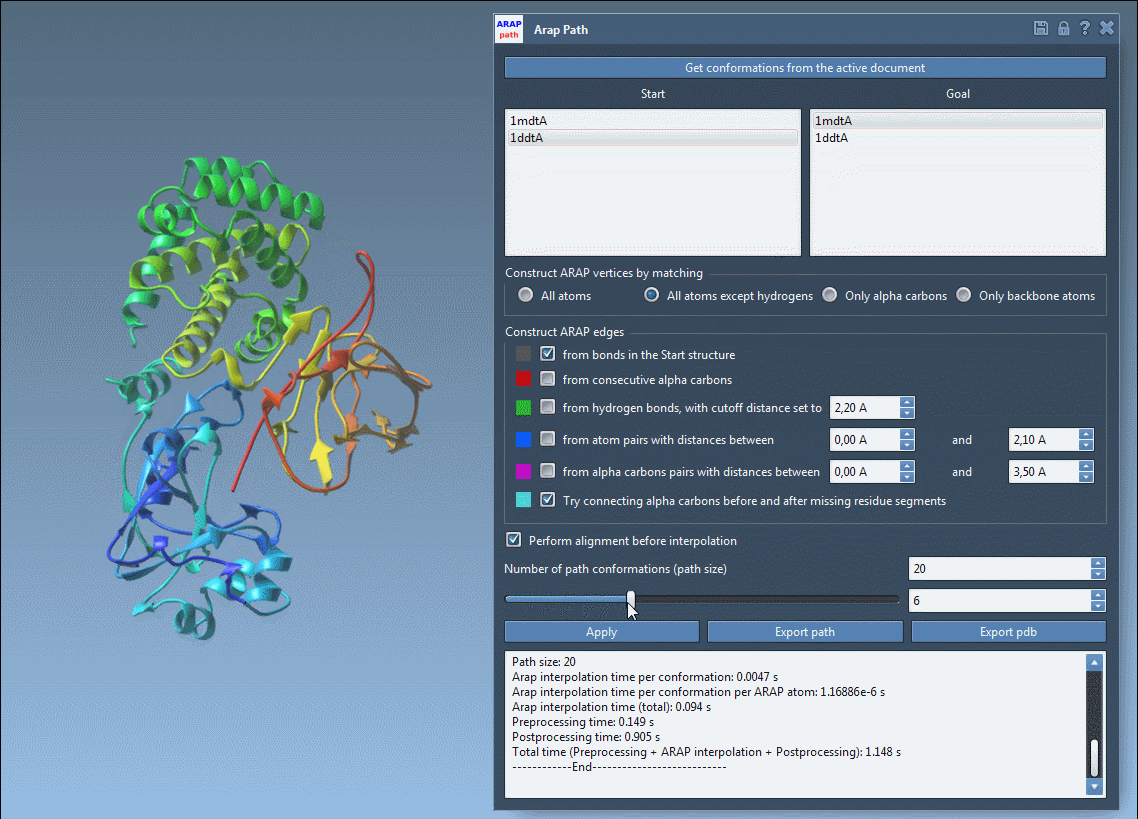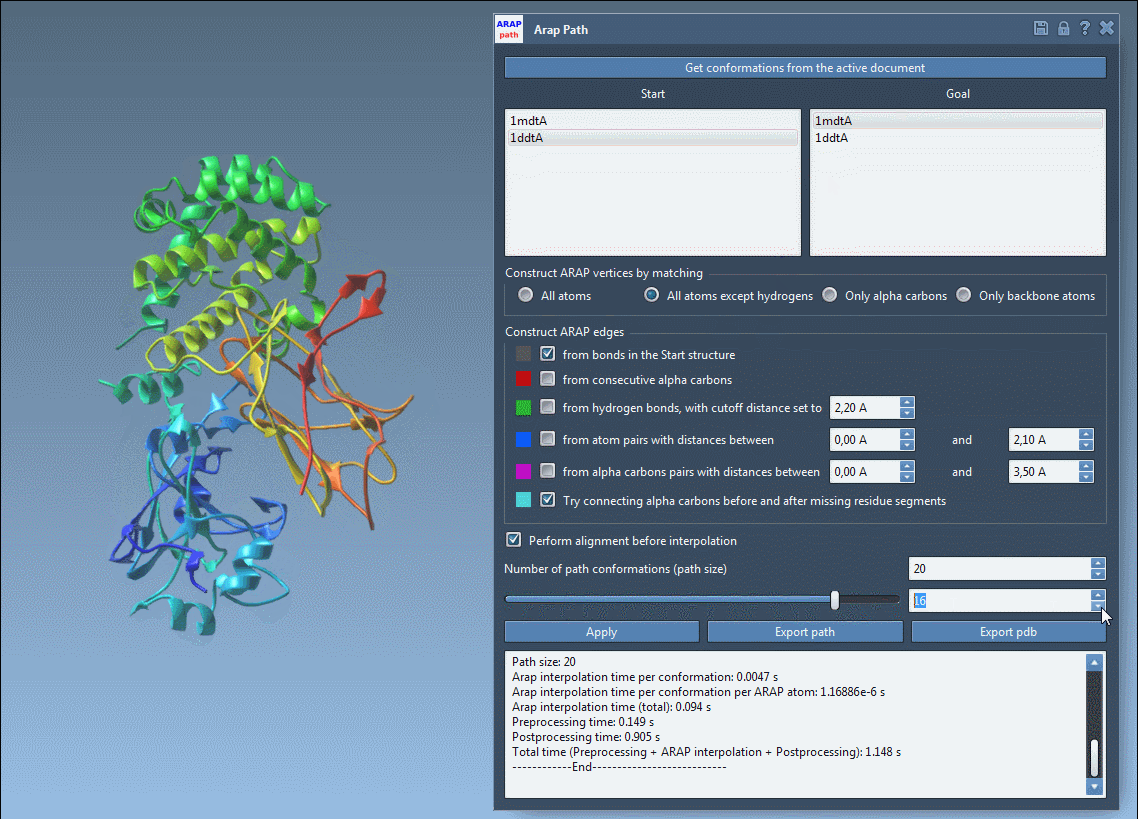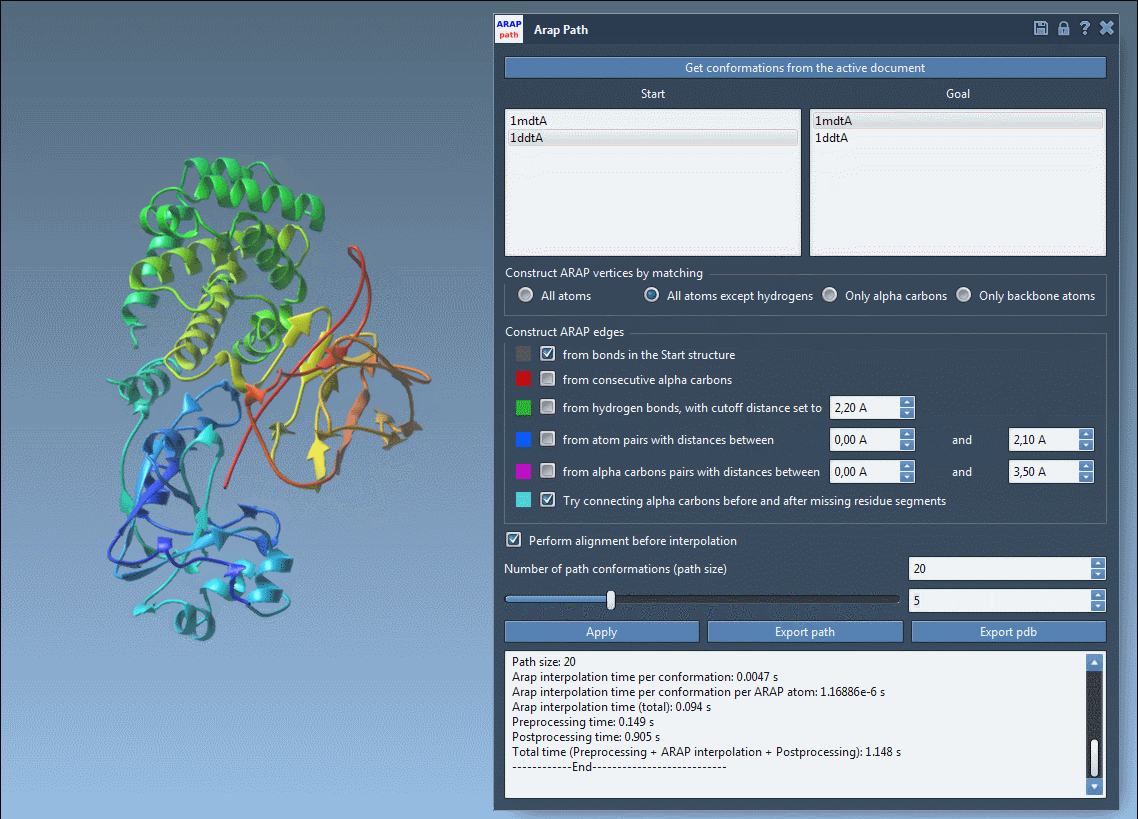
This interactive animation enables you to quickly evaluate the plausibility of the motion, providing immediate insights without running a full molecular dynamics simulation.
Export for Further Analysis
You can export the full path or save each conformation as an individual PDB file. These outputs can serve as inputs for:
- Umbrella sampling workflows (for example, in GROMACS)
- Refinement using steered MD or NEB methods
- Dimensionality reduction and clustering pipelines
Conclusion
If visualizing realistic transitions between protein conformations is a common step in your workflow, using the ARAP Interpolator can save you hours of preparation and preview time. It’s ideal for hypothesis generation, building simulations, or simply illustrating motion with biological significance.
You can learn more in the full documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON from samson-connect.net.