





In molecular modeling, identifying realistic transition paths between conformational states is often essential—whether you’re exploring ligand unbinding, folding pathways, or chemical reaction mechanisms. Yet, even with tools that generate trajectories, like linear interpolation or sampling-based methods, the paths can be rough and physically implausible.
That’s where the Parallel Nudged Elastic Band (P-NEB) method available in SAMSON becomes useful. This extension refines your initial transition path based on physical energy landscapes, correcting artificial distortions and revealing more realistic intermediate states.
💡 When should you use P-NEB?
Imagine you’ve generated a rough path from a bound to unbound ligand state using docking or a path generation tool like the Ligand Path Finder. On inspecting the trajectory, you notice irregular jumps or unlikely torsions. These issues can interfere with interpretation or further modeling (e.g., free energy calculations).
By applying P-NEB, you can optimize these paths using force fields and a minimization engine (such as FIRE), enforcing more physically meaningful movement. Here’s how SAMSON simplifies the workflow.
🔧 Getting Started with P-NEB
After installing the P-NEB Extension and the FIRE minimizer, you can either generate a path from existing conformations or download pre-built examples such as the Thiodigalactosid unbinding trajectory from this link:

To launch P-NEB, go to Home > Apps > All > P-NEB or use the search function.
⚙️ Recommended Settings
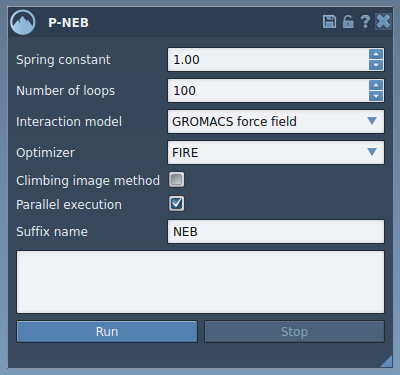
The P-NEB interface allows for adjustments, but a solid starting setup includes:
- Spring constant:
1.00 - Number of loops:
100 - Interaction model:
Universal Force Field - Optimizer:
FIRE - Climbing image: optional (to locate saddle points)
- Parallel execution: checked (for faster performance)
🧪 Run the Optimization
In the Document view, select the path node you’d like to optimize and hit Run in the app. If you’re prompted about bonds during the force field setup, choose to use existing bonds.
The system will optimize the path by minimizing the energy of each intermediate conformation while maintaining regular spacing. You can monitor progress in the status bar at the bottom of the SAMSON interface.


🔍 Review the Results
When the computation finishes, a new path labeled with your chosen suffix (e.g., “_NEB”) will appear in the document. You can double-click it to animate the transition or inspect conformations using the Inspector.
For users interested in refining atomic-level animations or building realistic reaction coordinates, this step provides a smoother trajectory that is compatible with downstream applications.
✨ Final Thoughts
Refining your molecular trajectories with the P-NEB app helps reveal more physically accurate transition mechanisms and plots a better foundation for interpretable simulations, from dynamics to thermodynamics.
To explore the full tutorial, visit the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.