





When optimizing molecular transition paths, one crucial decision can save you a significant amount of time and computational effort: choosing the right input format. In SAMSON, the P-NEB method allows you to refine ligand unbinding pathways (or any transition between molecular states), but how you go about it can make or break your workflow efficiency.
Paths vs. Conformations: What Makes the Difference?
SAMSON users often work with two kinds of nodes when calculating transition paths:
- Conformations: Individual snapshots of atomic positions, often saved manually or through computational methods.
- Paths: Trajectories composed of multiple atomic states animated over time, typically generated by simulation tools.
Both can be used as inputs for the P-NEB extension. However, it’s important to know that running P-NEB on a set of conformations takes significantly longer than applying it directly to a path representing the same transition steps.
Prefer Paths When You Can
The time difference comes from how SAMSON handles data internally. When conformations are used, SAMSON must interpret, interpolate, and manage several independent molecular snapshots. In contrast, paths are inherently sequential and continuous, streamlining the optimization process.
That’s why the documentation suggests favoring paths. If you currently have conformations but want faster results, you can easily convert them. Just follow this simple conversion:
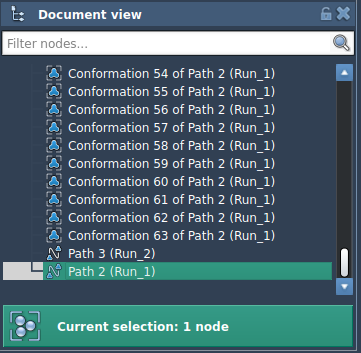
- Select the desired conformations in the Document view.
- Right-click and choose Conformation > Create path from conformations.
This new path can now be used as the input for P-NEB, significantly improving performance while preserving accuracy.
When You Might Still Use Conformations
There are still cases where conformations might be the better choice:
- If you’re comparing various hypothetical intermediate structures prior to committing to a complete path.
- When you’re manually generating transitions and want static control over each step.
But for most users aiming for optimized transition paths—particularly in tasks like ligand unbinding trajectories—it’s usually better to work with paths.
Visualizing the Workflow
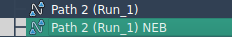
Whether you’re working with Zinc unbinding models or large-scale protein-ligand complexes, SAMSON makes it possible to visualize progress at each step. Here’s how a typical P-NEB session looks with paths:

After running the simulation, you’ll get a new, optimized path that can be examined with the Inspector, animated directly in the viewer, or exported for further use.
Try This in Your Next Project
If you’ve been using conformations by default, now’s a great time to revisit your workflow. The combination of smoother performance and easier animation control makes working with paths worthwhile for nearly all transition path optimizations.
To learn more and follow the full tutorial, check the official SAMSON P-NEB documentation here.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.