





One of the challenges molecular modelers often face when exploring chemical space is deciding where to modify a molecule. Minimal changes at the right location can significantly influence a molecule’s properties—whether you’re optimizing binding affinity, altering pharmacokinetics, or reducing toxicity. But identifying those positions systematically can be tricky. Fortunately, the SMILES Manager extension in SAMSON provides a highly visual and intuitive interface to address this issue using SMARTS-based pattern searching.
In this blog post, we’ll walk you through a self-contained feature of the SMILES Manager that lets you select specific structural patterns from a molecule—such as aromatic carbon atoms—and prepare them for substitution or extension. This allows you to set up positional analogue scanning experiments with just a few clicks, directly in SAMSON.
Why pattern selection matters
Before diving into any substitution, it’s crucial to pinpoint chemically meaningful and diverse locations within a molecule that may tolerate change or significantly impact activity. This is where SMARTS (SMILES Arbitrary Target Specification) shines. It’s a powerful way to describe molecular fragments using symbolic rules. For example, the SMARTS code [cH] lets you select all aromatic carbon atoms with a hydrogen—potentially excellent candidates for substitution.
Selecting a pattern step-by-step
Assuming you’ve already loaded your molecule into SAMSON, either by SMILES code or selection in the interface, here’s how the pattern selection works:
- In the SMILES Manager, locate the field that accepts SMARTS input.
- Enter the desired SMARTS pattern. For example, try
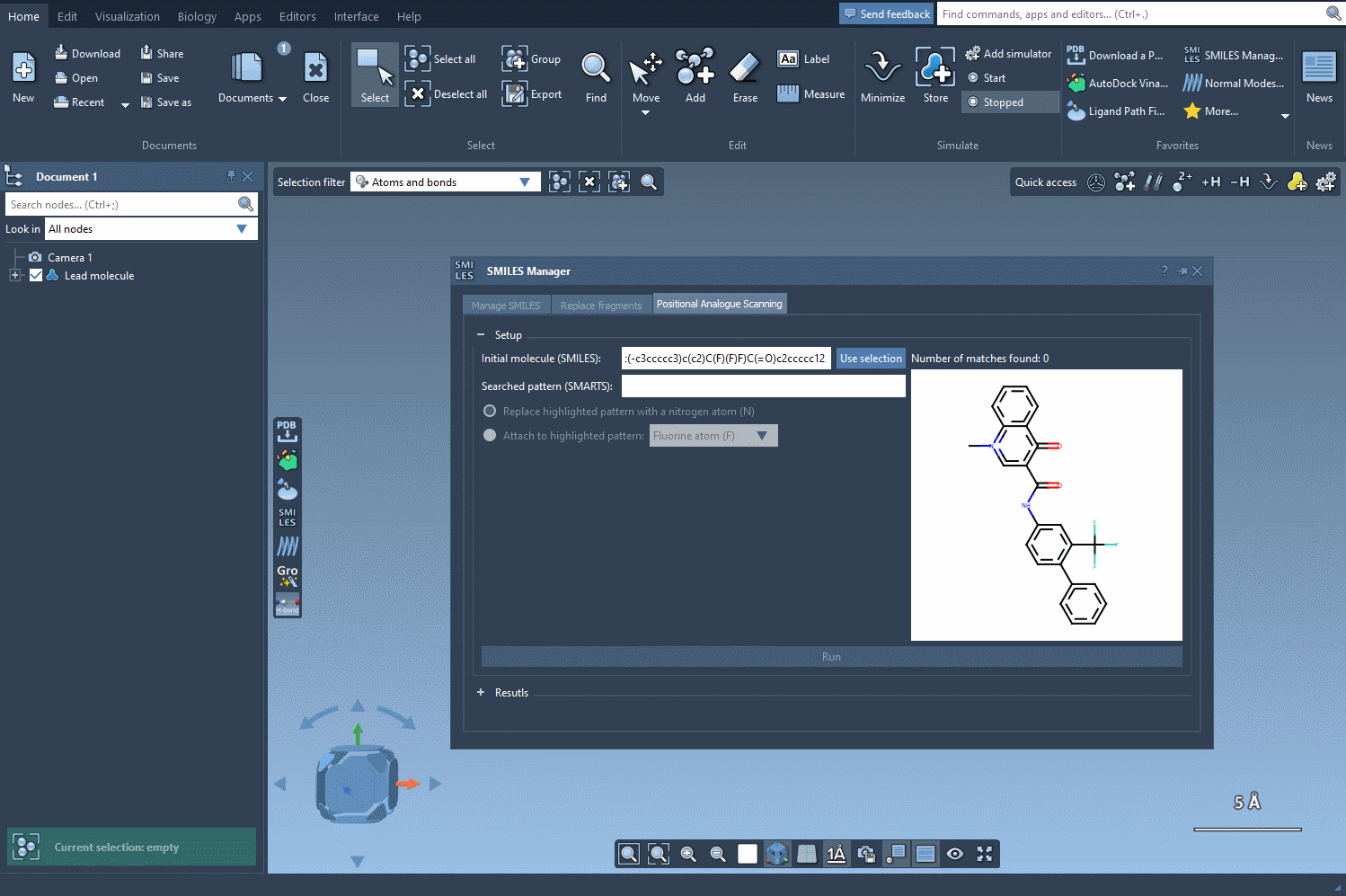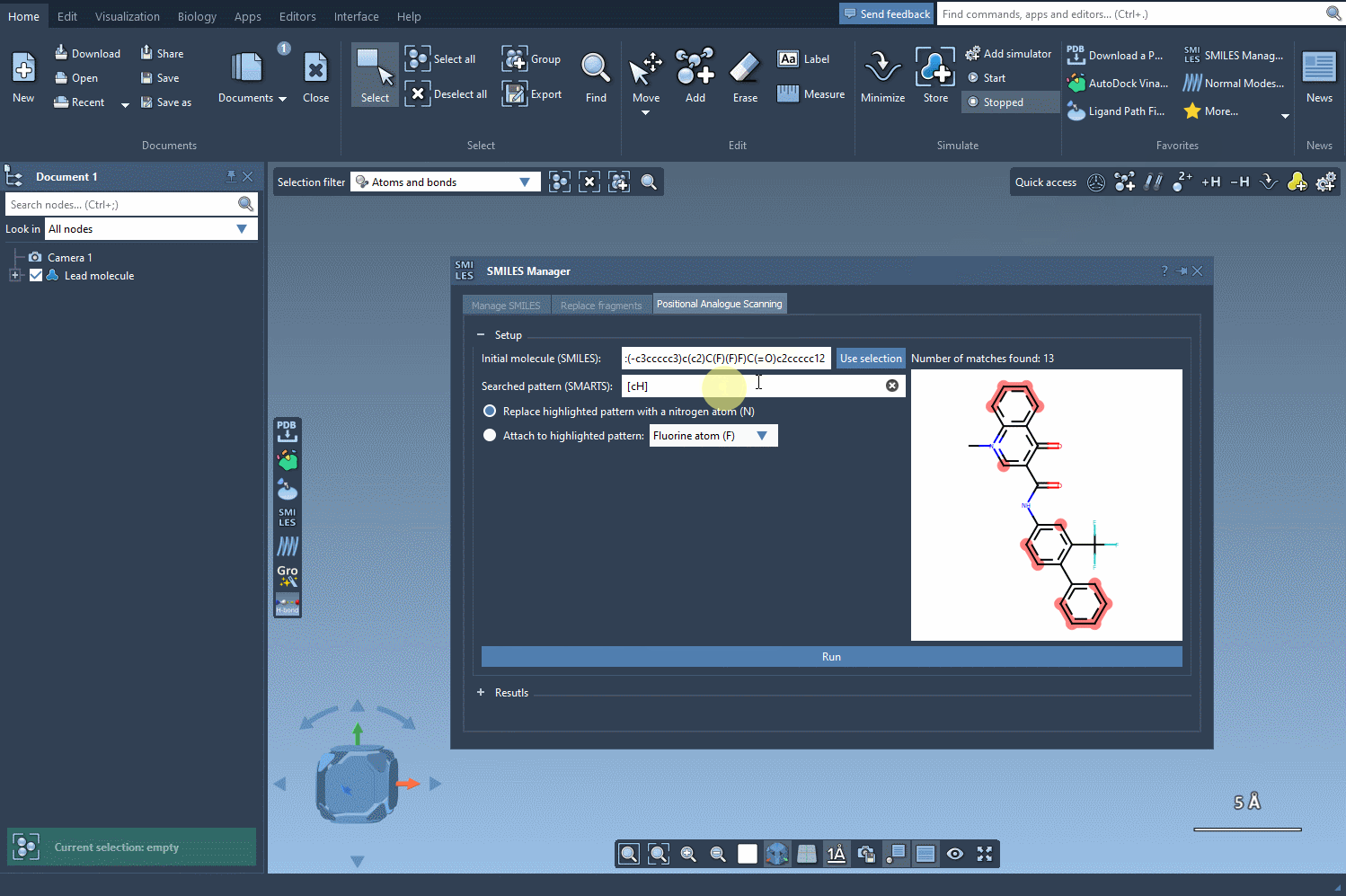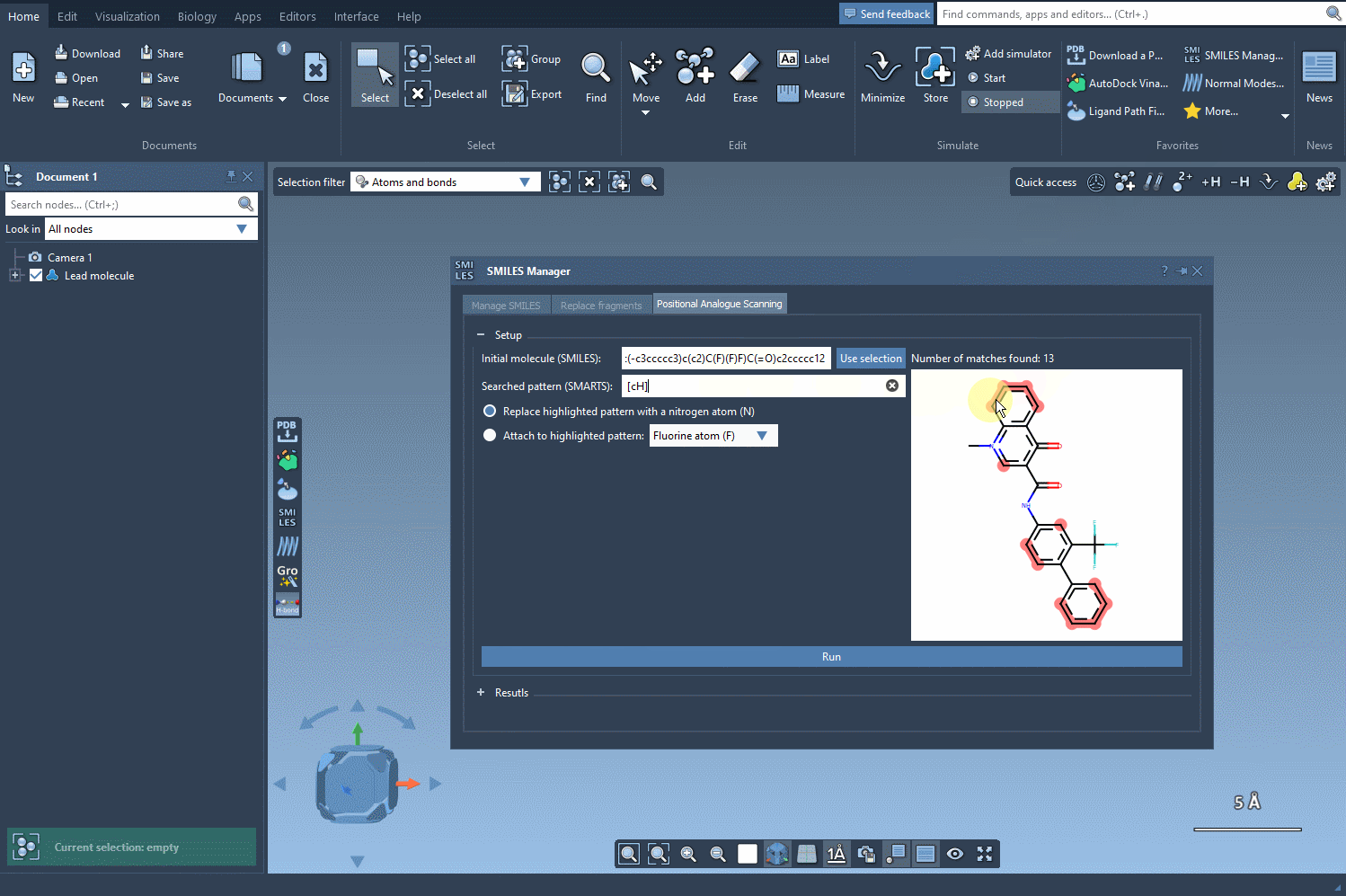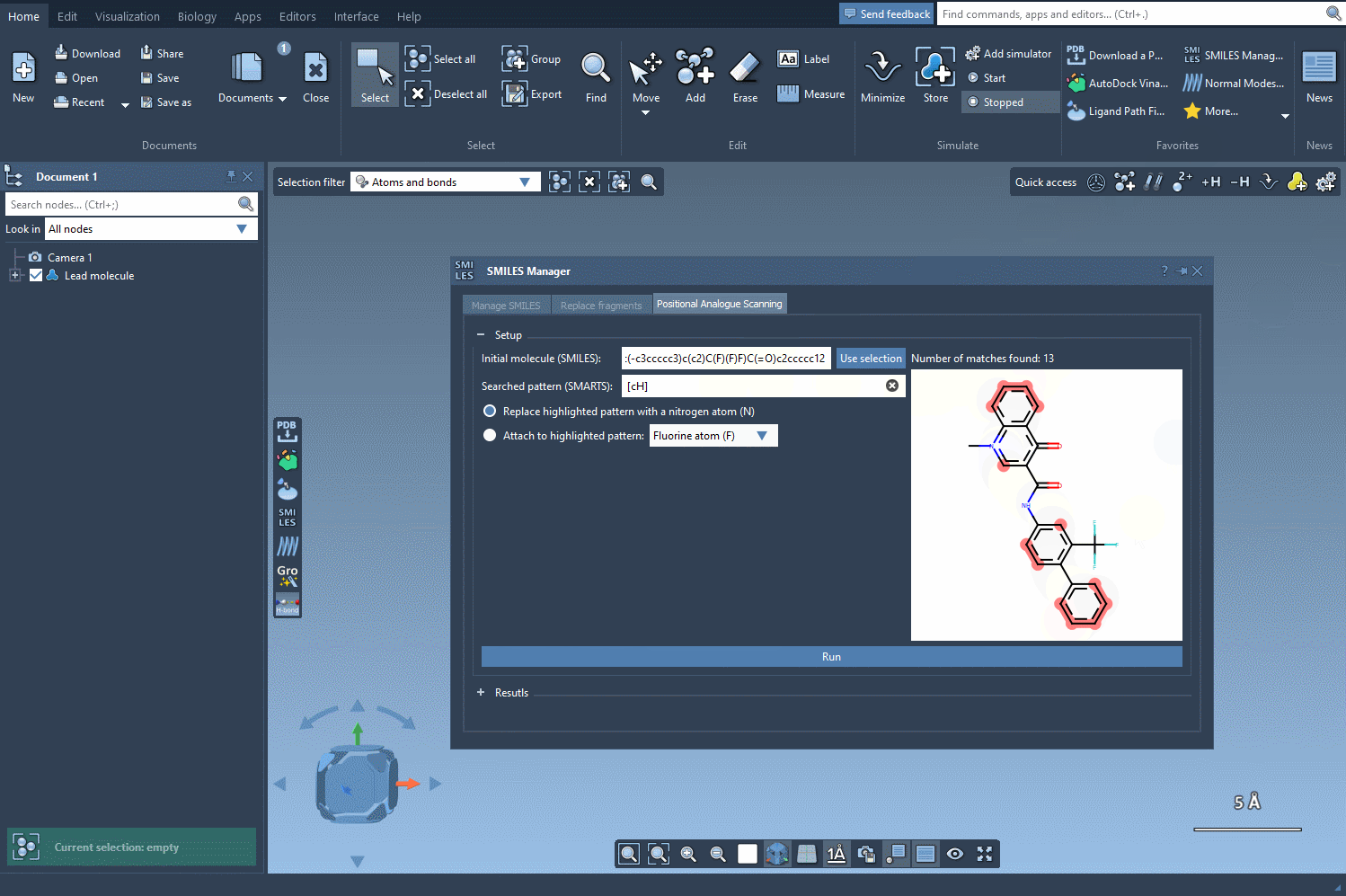
[cH]to find unsubstituted aromatic carbons. - The matched atoms will be instantly highlighted in the structure, providing immediate visual feedback on which parts of the molecule are being selected.
This live SMARTS pattern matching helps guide your decision-making visually—which is especially helpful in more complex molecules. You instantly see how many atoms match your pattern and where they are. This improves confidence before moving on to modifications like atom replacement or group attachment.
Tips for effective pattern use
- Experiment with different SMARTS patterns—SAMSON will update the highlights in real time, helping you learn as you go.
- Use specific SMARTS to target functional groups, heteroatoms, or atoms with defined hybridization (e.g.,
[N+],[O-]). - If no atoms are highlighted, double-check the pattern syntax. The real-time feedback is highly useful for debugging.
By integrating pattern-based scanning into your workflow, you’re no longer guessing where to make modifications: you’re building a focused, hypothesis-driven list of changes to test.
Once you’ve selected the pattern, the SMILES Manager makes it easy to automatically generate analogues by replacing or attaching atoms/groups to the selected pattern. You can then go on to evaluate these analogues using other tools like docking with Autodock Vina Extended—all within the same platform.
Learn more about setting up positional analogue scans and see additional examples and visuals in the full documentation here: Official SMILES Manager Tutorial.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON here: https://www.samson-connect.net.