





Understanding the dynamics of biological macromolecules like proteins is crucial for many molecular modeling applications, from drug design to structural biology. One common pain point? Visualizing and simulating conformational transitions between different functional states of a protein — especially when structural variations exist between those states.
In this post, we’ll look at how the opening motion of the SARS-CoV-2 spike protein was modeled within SAMSON, the integrative molecular design platform, and how this approach can help you simulate similar transitions in your own molecular systems—even when your starting and end structures differ slightly.
Why this matters
The SARS-CoV-2 spike protein (S protein) is a trimeric transmembrane protein that enables the virus to enter host cells by recognizing and binding to the ACE2 receptor. For this to happen, the spike undergoes a conformational change from a closed (inactive) state to an open (active) state. Understanding and visualizing this transition can provide valuable insight into viral infectivity and potential sites for therapeutic intervention.
But there’s a catch: the closed and open structures used (PDB 6VXX and 6VYB, respectively) differ in their number of residues, making the modeling task more complex for researchers who rely on consistent residue mapping through trajectories.
How the motion was modeled
Here’s how the team behind SAMSON tackled this:
- Preparation: Both PDB structures (6VXX for closed and 6VYB for open) were preprocessed—bond orders of sugars were adjusted using a Python script, and hydrogens were added.
- Minimization: The structures were energy-minimized to stabilize them for interpolation.
- ARAP Interpolation: The As-Rigid-As-Possible (ARAP) Interpolation Path module was used to create an initial path between the two states.
- Residue mismatch workaround: Since 6VXX and 6VYB differ in residues, an intermediate structure from the ARAP-generated path was selected and minimized to serve as a better-matching closed conformation.
- Refined interpolation: The interpolation was re-run using the adjusted closed structure as the target.
- Optimization: The path was further refined using the P-NEB (Parallel Nudged Elastic Band) module to improve the physical plausibility of the transition.
What you can get
The result is a high-quality, shareable animation showing the transition from closed to open states of the spike protein—a valuable educational and research tool that can help scientists understand how receptor recognition and membrane fusion might occur. Here’s a sample, displayed from the side view:
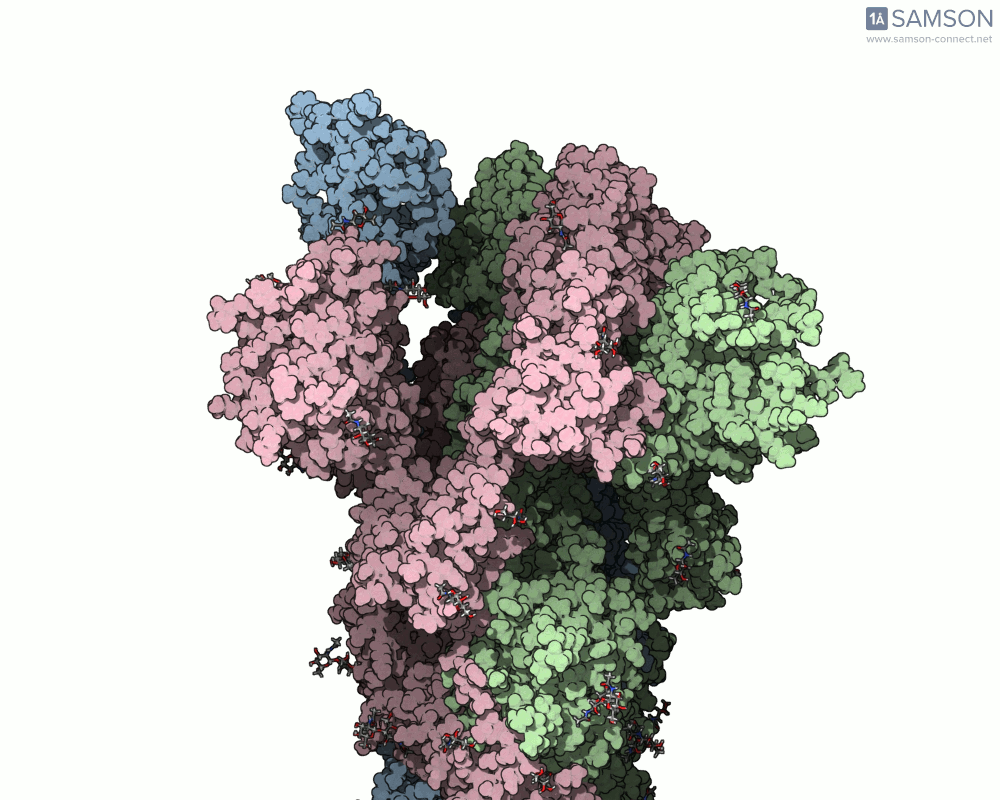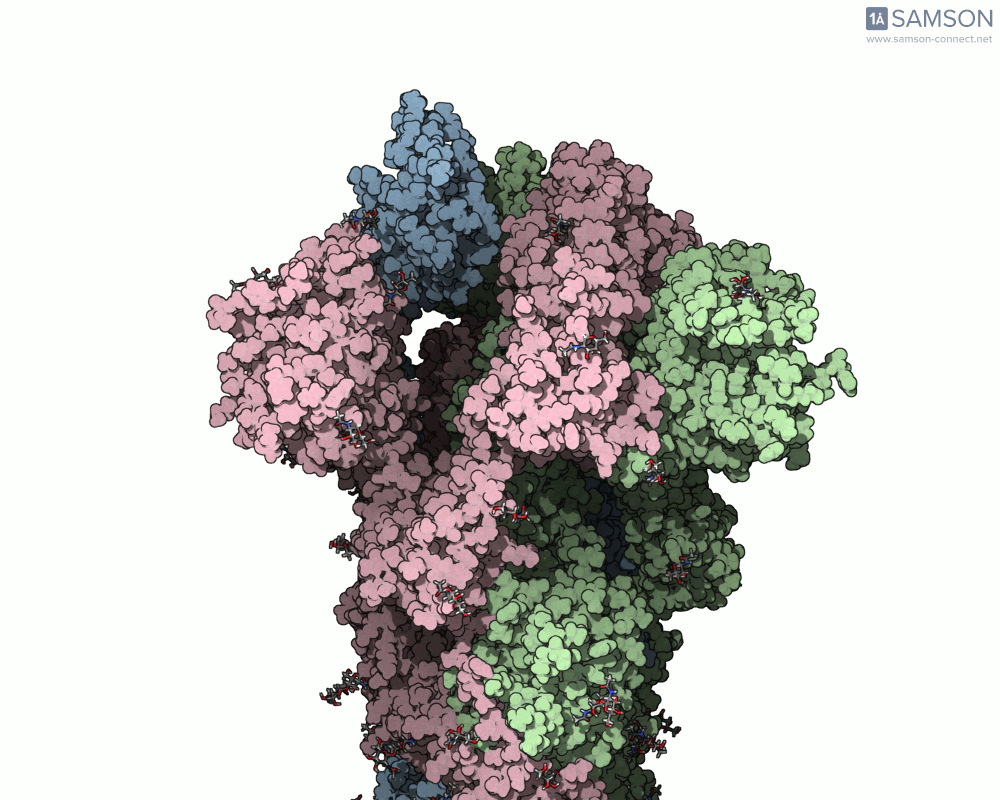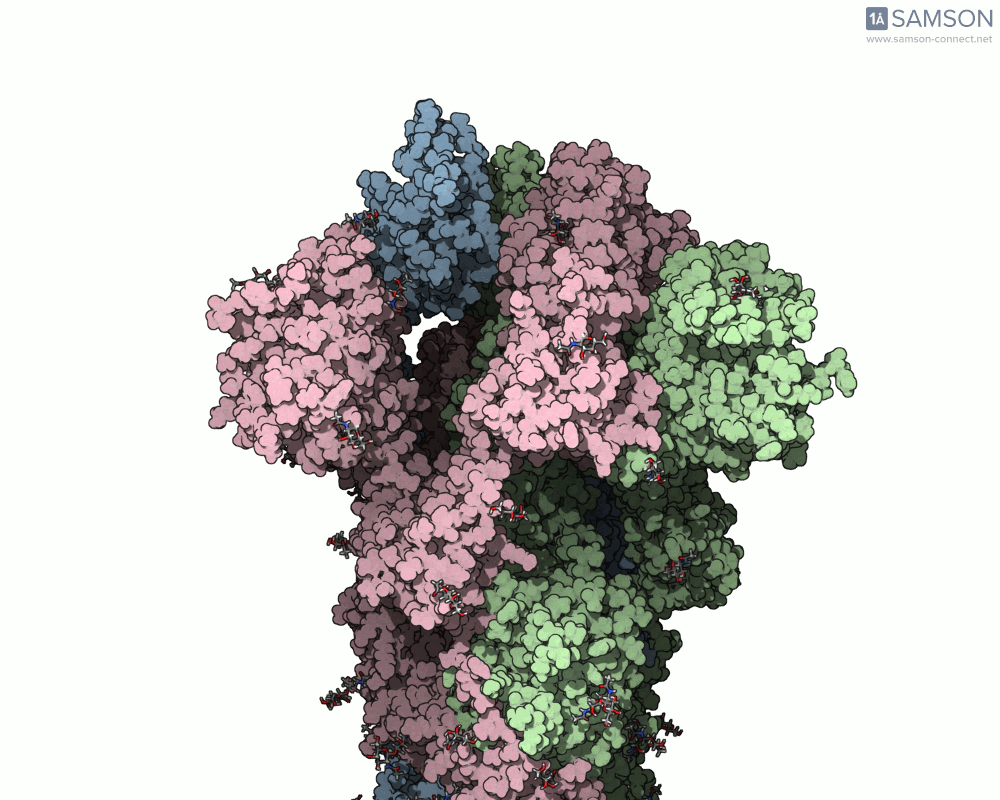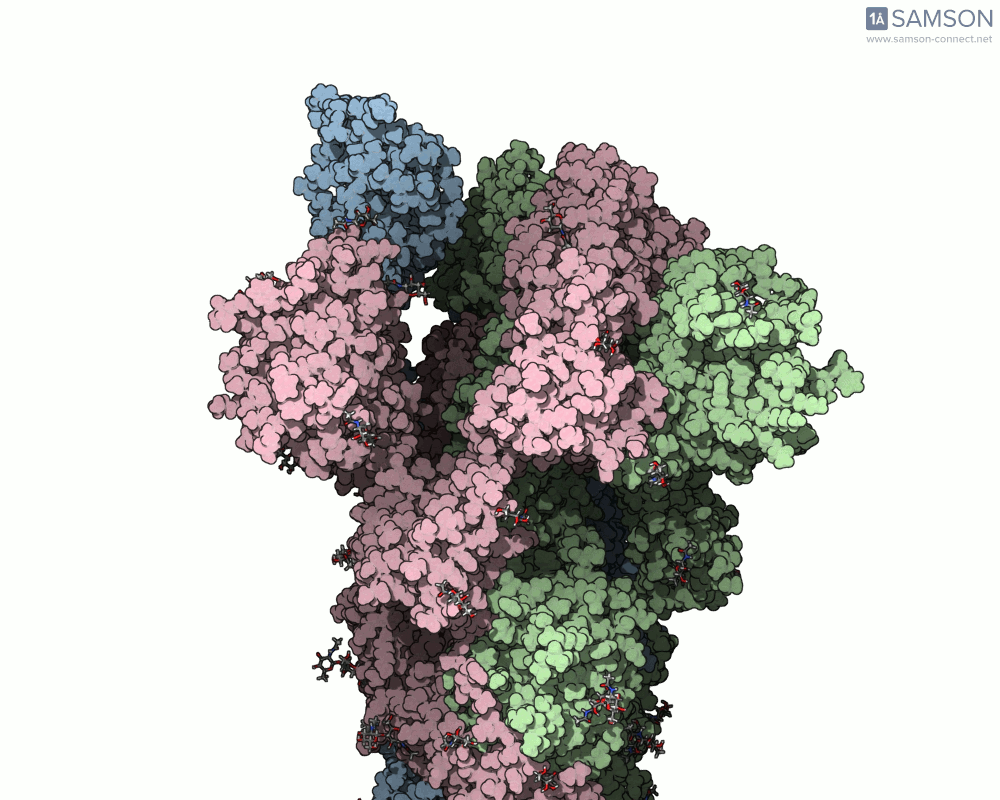
You can also download the trajectory files (PDB or SAMSON formats) and adapt them for further study, reinterpret them under different biophysical assumptions, or combine them with mutational variants of the spike.
Replicating this for your own molecules
This workflow isn’t exclusive to viral spikes—you can use the same approach for other flexible biomolecules where you have (or can model) two endpoint conformations, even if they don’t match exactly in residues. The combined use of ARAP for fast interpolation and P-NEB for refinement makes it practical on standard laptops, letting you explore conformational transitions in days rather than weeks.
Being able to visualize these motion paths helps in many areas: identifying intermediate conformations, exploring potential allosteric sites, or generating input for docking or molecular dynamics simulations.
To learn more about this workflow and get access to everything shown here, visit the original documentation: Computing the Opening Motion of the SARS-CoV-2 Spike.
SAMSON and all SAMSON Extensions are free for non-commercial use. To start modeling, download SAMSON from https://www.samson-connect.net.