





When working on lead optimization or structure-activity relationship (SAR) studies, chemists often ask: what happens if I substitute this atom or group at a specific position in my molecule? Answering this question manually for many analogs can be a tedious and error-prone process.
Fortunately, SAMSON’s SMILES Manager introduces a straightforward solution through positional analogue scanning—allowing researchers to quickly generate molecule variants based on search patterns like [cH] to define substitution targets.
This post walks you through a key step in this useful method: how to define and highlight a specific atom or substructure pattern using SMARTS. This step helps researchers focus the exploration of their compound space to chemically meaningful substitutions by targeting parts of a molecule such as an aromatic carbon, a nitrogen atom, or a methyl group.
Why SMARTS Pattern Matching Matters
Imagine working on a molecule with multiple aromatic rings. You want to test substitutions only on hydrogen-bearing aromatic carbons (often hotspots for fine-tuning properties like solubility or activity). But how do you programmatically identify those positions?
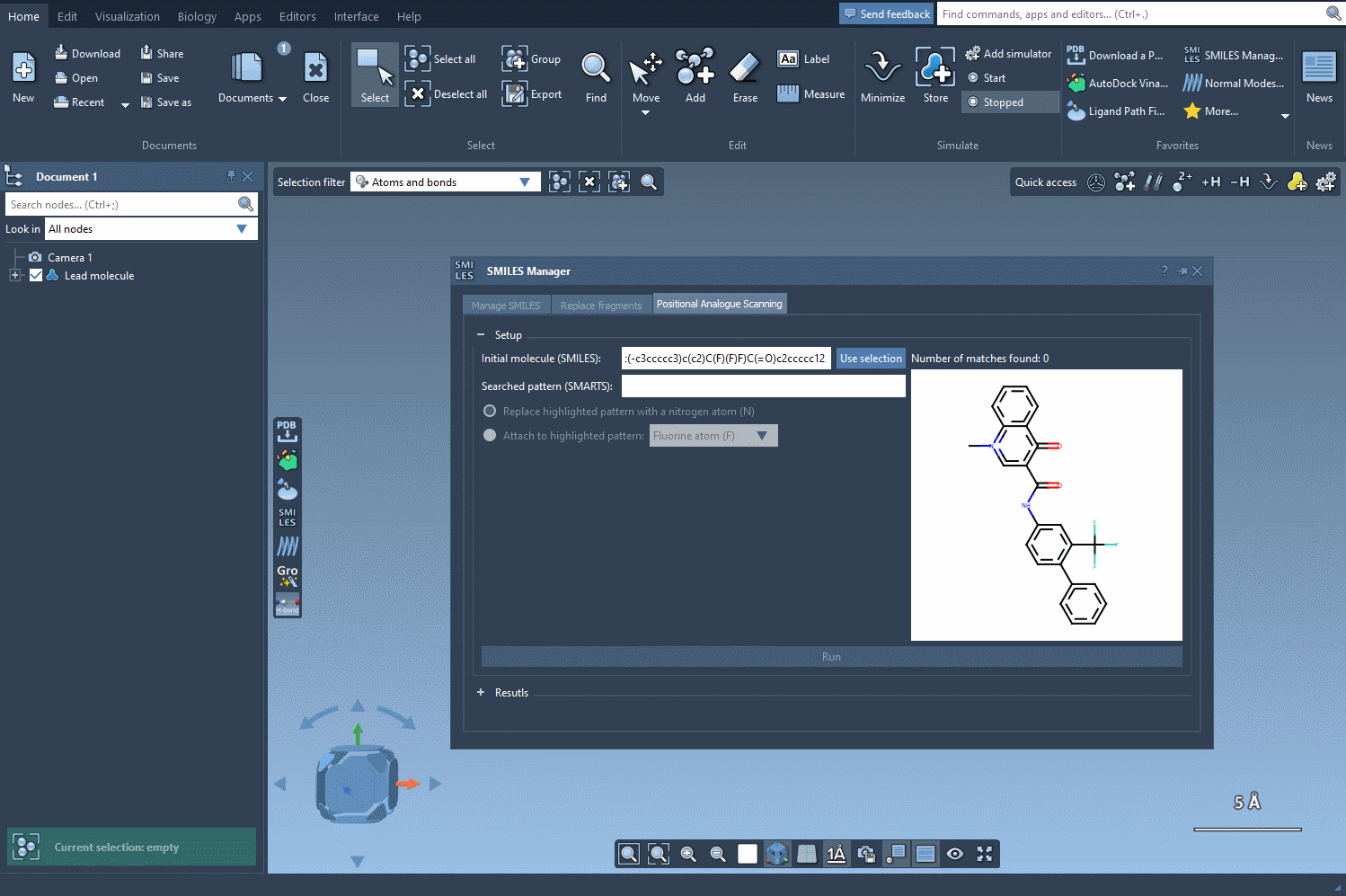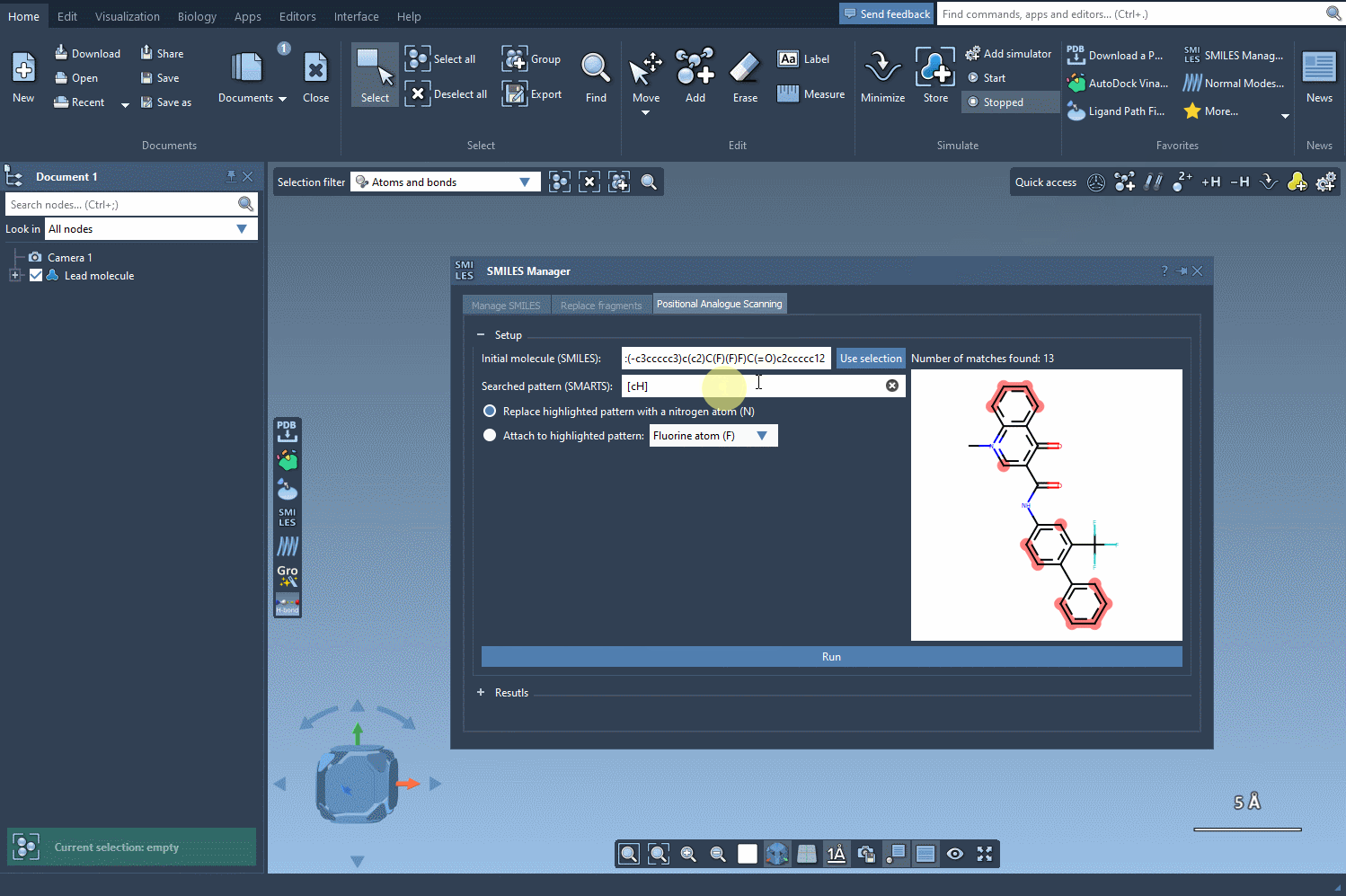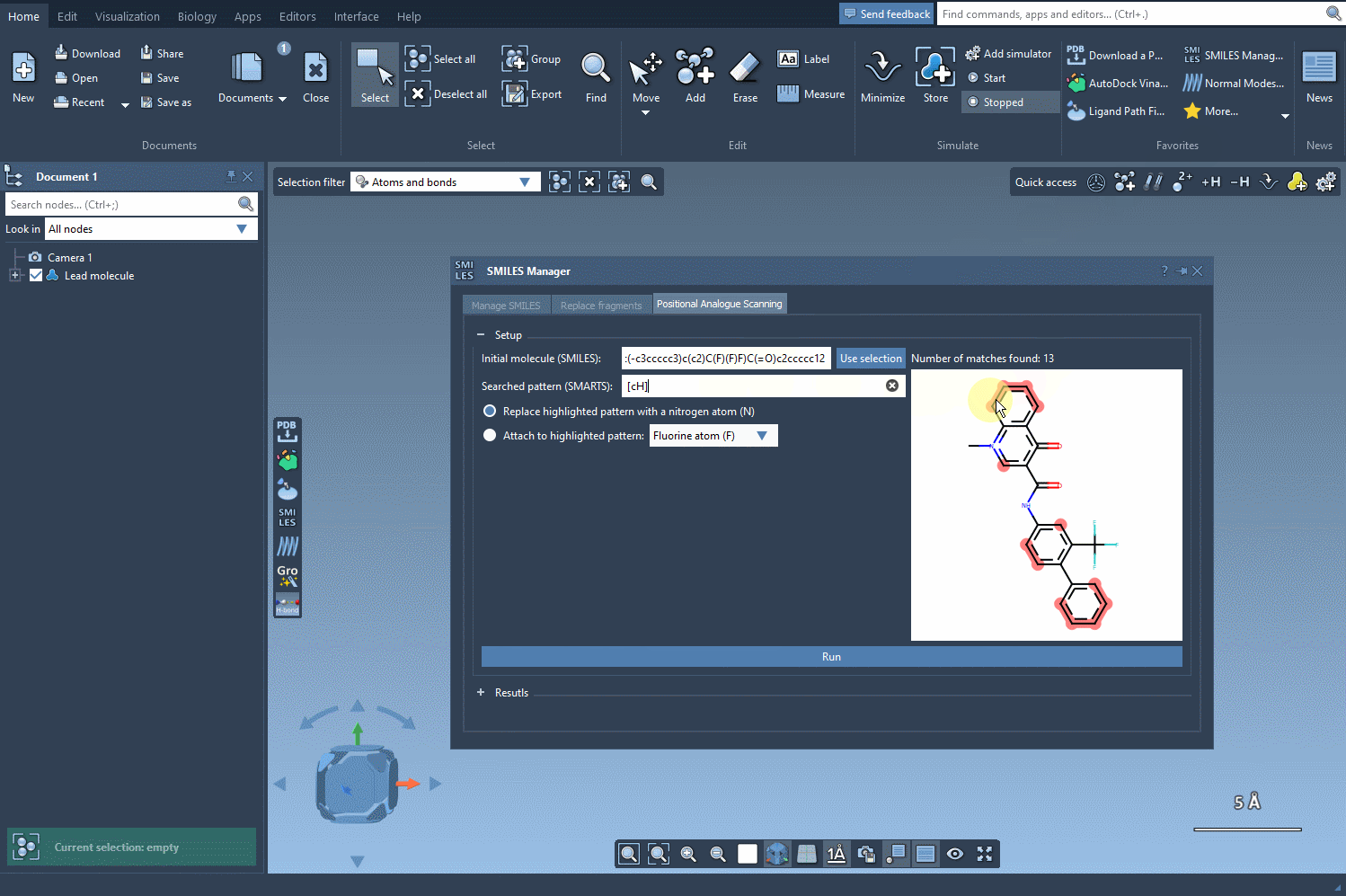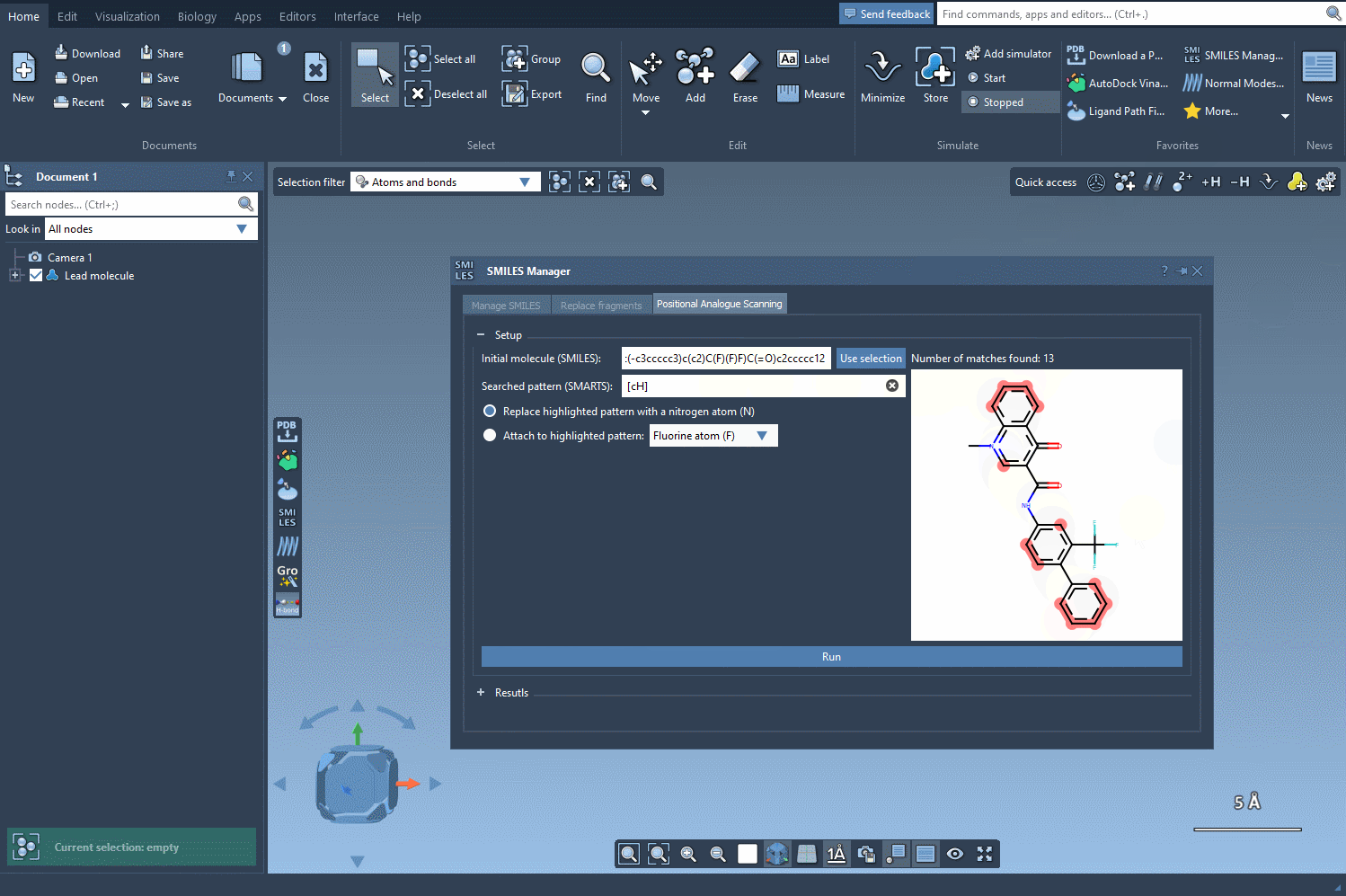
That’s where SMARTS (SMiles ARbitrary Target Specification) comes in. Using a pattern like [cH], you can instruct the SMILES Manager to find and visually highlight all positions in your molecule that contain aromatic carbon atoms with a single hydrogen attached. These are common substitution targets in SAR design.
How It’s Done in SAMSON
Once you’ve selected or input your starting molecule in SAMSON:
- Navigate to the Search pattern field in the SMILES Manager interface.
- Input your SMARTS pattern, for example:
[cH]. - The software then scans your molecule, detects matches to this pattern, and highlights them directly in the molecule viewer.
This step provides immediate visual feedback, ensuring that your defined pattern is matching the intended atoms only.
By visually confirming the intended pattern locations, you reduce the risk of off-target modifications and make sure your analogue library is chemically relevant and meaningful for downstream studies like docking or pharmacophore modeling.
Tips to Make the Most of It
- Use more specific SMARTS patterns to target substructures or functional groups. For example,
[c:1][nH]matches specific aromatic carbon-nitrogen pairs. - If your initial pattern highlights too many atoms, consider refining it to avoid unnecessary substitutions.
- SAMSON’s interface allows dynamic updates; you can tweak patterns and rerun without restarting your setup.
What’s Next?
After defining your target pattern, SAMSON makes it easy to replace or attach new atoms or functional groups, generate analogue SMILES, visualize them in 2D, and convert them into 3D structures. This makes pattern-based analogue generation fast, flexible, and accessible—even to non-programmers in drug design or computational chemistry.
For the full tutorial and step-by-step guidance, visit the official documentation:
https://documentation.samson-connect.net/tutorials/smiles-manager/perform-positional-analogue-scanning-using-the-smiles-manager-element/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.