





One common challenge in molecular modeling is verifying that a protein structure is ready for simulation. Even with high-quality data or carefully built homology models, strained residues can sometimes go unnoticed. These outliers can result in unstable simulations and inaccurate predictions.
This is where the Ramachandran plot shines as an essential validation step for protein modeling workflows. With the Interactive Ramachandran Plot extension in SAMSON, you can not only visualize problematic residues—but also fix them, all from the same plot.
What Makes a Residue Strained?
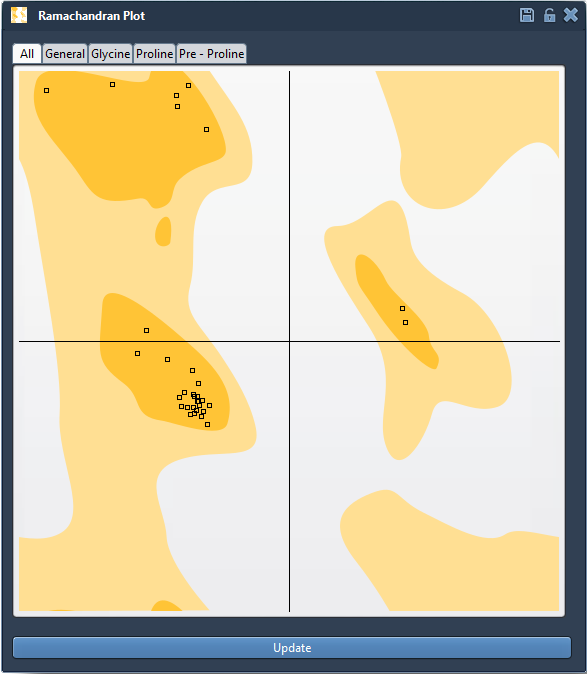
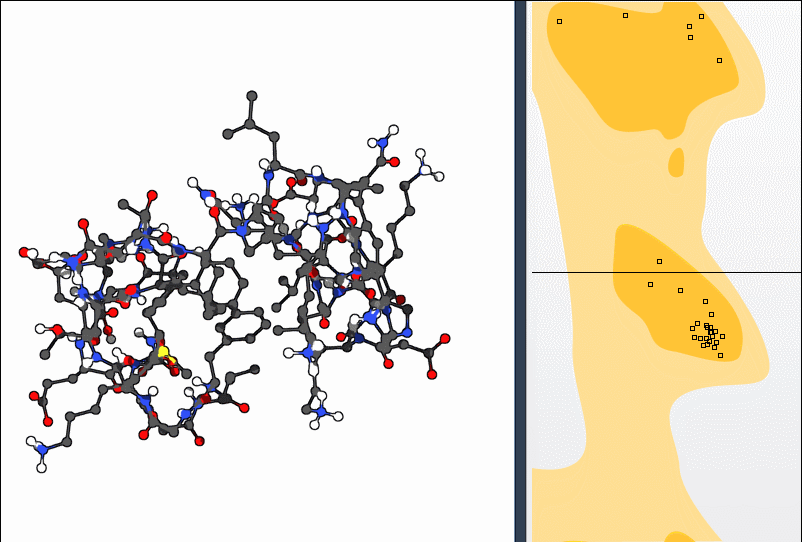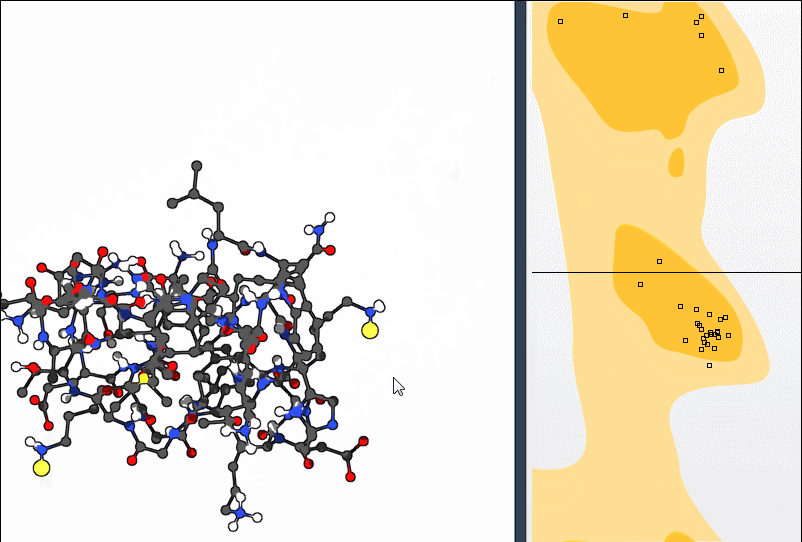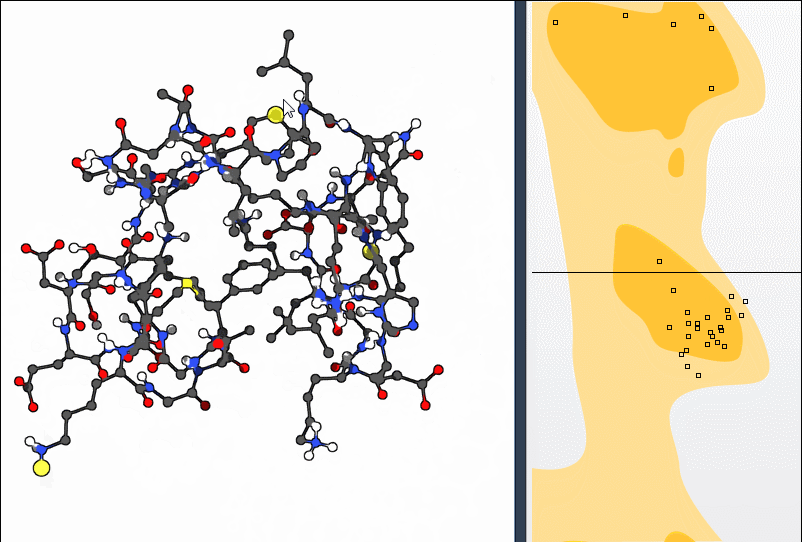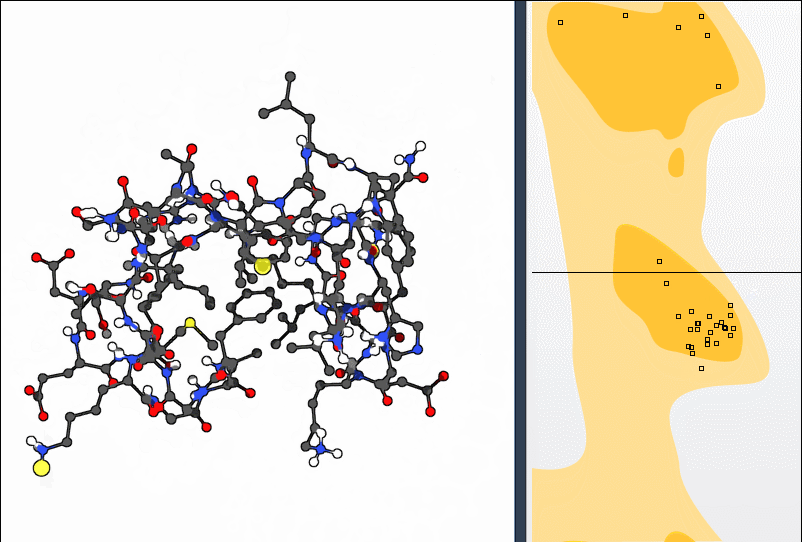
Every amino acid residue in a protein backbone has two key dihedral angles: φ (phi) and ψ (psi). Because of steric hindrance and energetic constraints, only certain combinations are physically allowed. The Ramachandran plot maps these combinations:
- Yellow areas indicate favored conformations.
- White regions correspond to disallowed or strained conformations.
How to Catch and Edit Outliers
Once your protein is loaded into SAMSON (for instance, using PDB ID 1YRF), launch the Ramachandran Plot App via:
Home > Apps > Biology > Ramachandran Plot
Click the Update button to generate the plot. Each point on the plot represents a residue. If any of them fall outside favored regions, it’s worth investigating.
Clicking on a specific point selects the corresponding residue in the 3D view and shows its exact φ and ψ values in the status bar:


Two Ways to Fix Dihedral Angles
If a residue is in an energetically unfavorable position, you can modify it directly from the plot in two intuitive ways:
1. Dragging Points on the Plot
- Click and drag a residue point to new φ/ψ values.
- The 3D structure updates in real time.
- Use Ctrl / Cmd + Z to quickly undo any changes.
2. Using the Twister Editor
You can also manipulate the structure in 3D using the Twister editor from the left-hand menu:
This method helps you see structural consequences while refining local geometry.
Why This Matters
Validating and adjusting backbone angles before simulation helps avoid crashes or artifacts due to impossible conformations. It’s particularly helpful when working with:
- Homology models
- Poorly resolved experimental structures
- Structures manually edited or repaired
In just a few clicks, you can resolve conformational outliers and prepare your system for downstream applications like normal mode analysis, docking, or molecular dynamics.
To learn more, visit the full tutorial at this page.
SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at https://www.samson-connect.net.