





When working with protein-ligand complexes in solution, interpreting ambiguous NMR signals can be tricky. The NMR2 extension in SAMSON helps resolve these ambiguities using spatial restraints to predict complex structures. But to benefit from the algorithm, the first step—defining your system correctly—is critical.
Many researchers spend significant time troubleshooting structure preparation because the algorithm’s accuracy depends on the correct identification of the protein, ligand, and nearby residues. Here’s a straightforward way to define the required structures in SAMSON to reliably kickstart your NMR2 analysis.
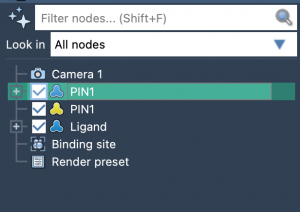
1. Use the Document View to Avoid Misclicks
The Document view in SAMSON is your central tool to select and define molecular components. Here, you can clearly see structural elements and select them with precision.
2. Assign Structures in the NMR2 Interface
Once your protein, ligand, and nearby residues are visible in the Document view, you can assign them in the NMR2 interface:
- Receptor: Click on the protein (e.g.,
PIN1) and hit Set. - Ligand: Select the ligand structure and click Set. This automatically renames atoms and adds pseudo-atoms, which will be referenced in distance restraints.
- Binding site residues: Select the predefined group or choose relevant residues and click Set. These likely host or neighbor methyl groups observed in the NMR experiment.
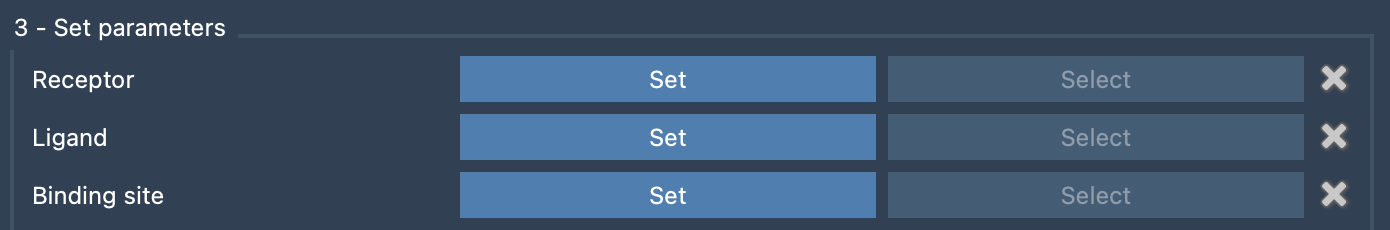
These three steps define the search space for methyl assignments and reduce errors caused by incorrect or missed atom selections.
3. Use Predefined Groups to Speed Up Setup
If you or your team frequently use a specific set of binding residues (e.g., LEU 61, LEU 122, MET 130, LEU 141), grouping them into a saved selection allows consistent reuse and saves time.
Tip
Even though this tutorial uses saved selections, you can always make your own by selecting residues in the Document view or using SAMSON’s Find command.
4. Why It Matters
Defining the structures carefully impacts more than aesthetics. The downstream prediction of ligand positions and methyl assignments relies on exact atom naming, consistent assignments, and valid spatial constraints. Even a small mismatch in pseudo-atom naming can cause the algorithm to skip restraints—or worse, accept invalid ones.
Practical Benefits
- Reduces trial-and-error iterations
- Improves reproducibility when using shared or published datasets
- Makes collaborative work easier by introducing consistent naming conventions
Understanding the structure setup stage is one of the most underappreciated yet impactful aspects of predicting protein-ligand binding via NMR signals.
Learn more in the full tutorial on NMR2: https://documentation.samson-connect.net/tutorials/nmr2/predicting-protein-ligand-complexes-using-nmr2/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net