





Understanding the structural transitions of viral proteins is key in drug discovery and vaccine development. One of the most critical mechanisms in SARS-CoV-2 infection is the conformational change of the spike (S) protein from a closed state to an open, receptor-binding state. For many molecular modelers, visualizing and simulating such complex protein transitions can be technically challenging and computationally intensive.
To support researchers and educators, SAMSON — the integrative molecular design platform — provides a practical, illustrated view of how the SARS-CoV-2 spike protein opens up to bind to the ACE2 receptor. This simulation illustrates a continuous motion trajectory between two experimentally determined states of the protein. The trajectory is computed in SAMSON using freely available tools and can be viewed in 3D, frame-by-frame, or as smooth animations. Here, we summarize how this complex protein motion was computed and how you can explore it.
From Closed to Open: A Molecular Journey
The transition from the spike’s closed (down) state to its open (up) state is biologically significant. It’s only in the open state that the spike can recognize and latch onto the ACE2 receptor on human cells, initiating infection. SAMSON’s computed trajectory highlights this conformational change in several intuitive visual formats:
- Side views allow you to observe the upward motion of part of the spike protein.
- Top-down views reveal the twisting and opening of the spike’s trimeric structure.
- Oblique angles give a spatial understanding of how one subunit rises while others stay at rest.
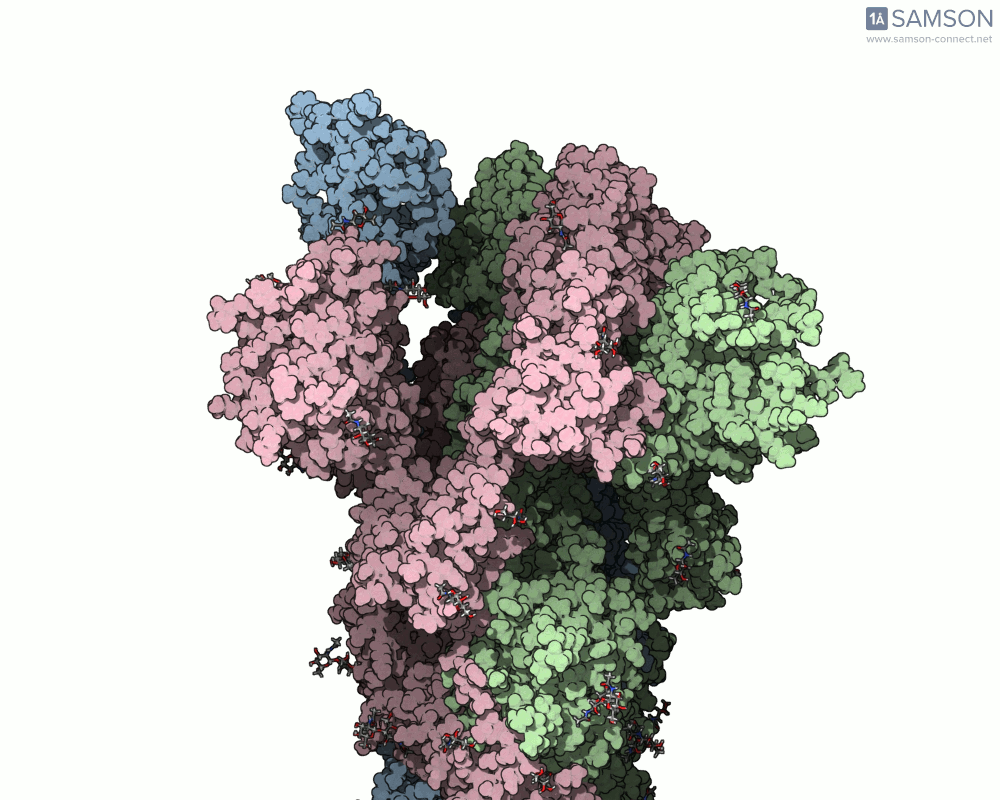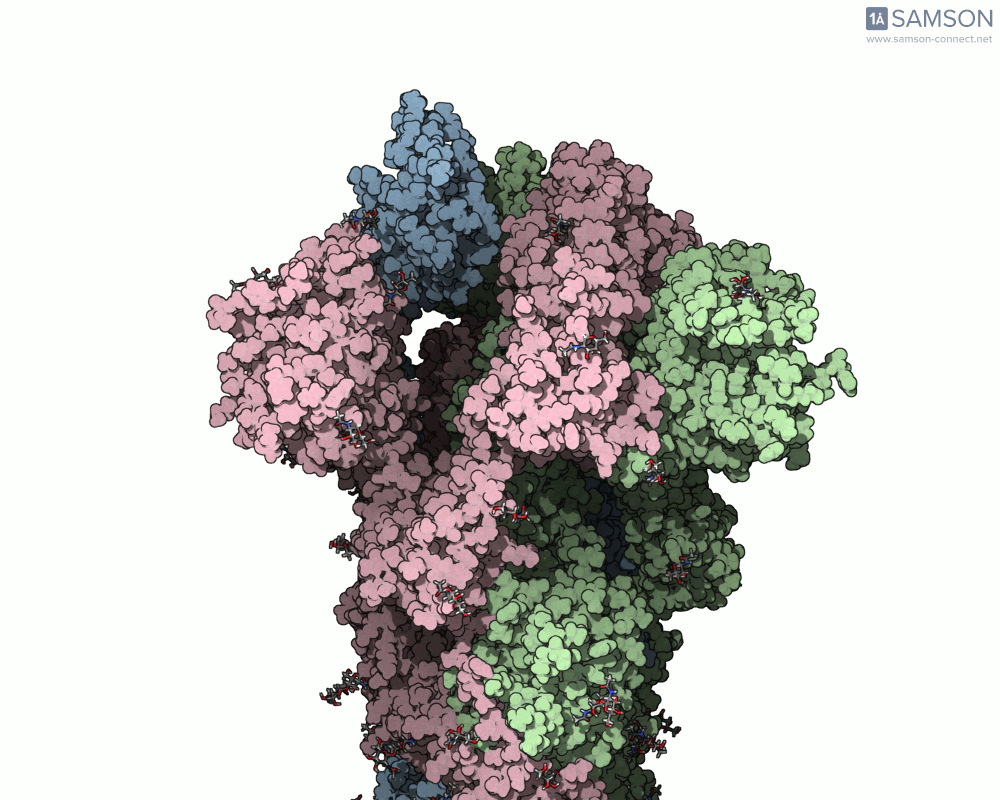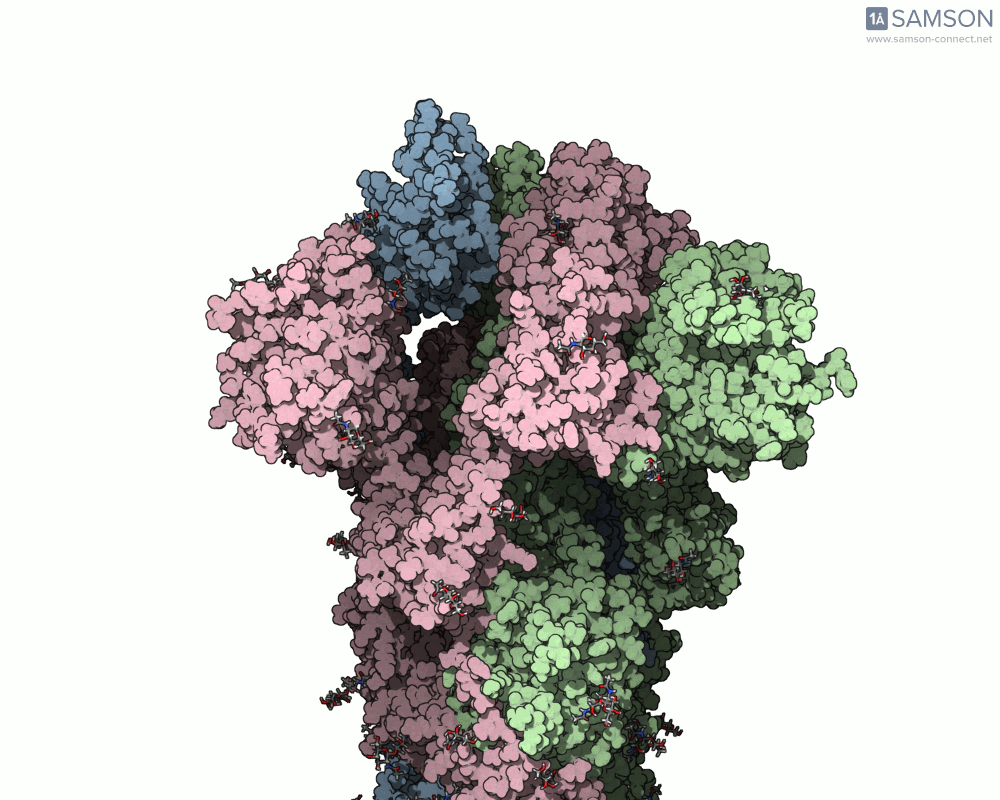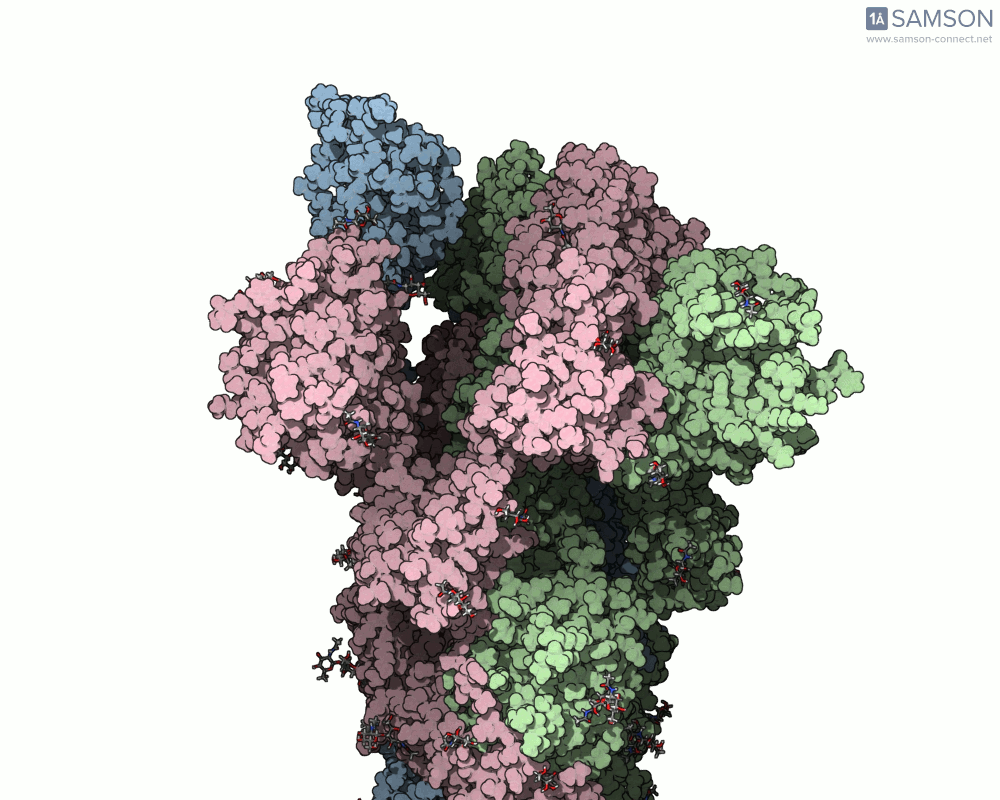
Side view of the spike in motion.
How the Spike Motion Was Computed
To generate this trajectory in SAMSON, the team started from two PDB structures:
Here’s a high-level overview of the workflow:
- Preprocessing: Bond orders in sugars were corrected using a Python script.
- Hydrogen atoms were added and structures were minimized.
- ARAP Interpolation Path Module produced a fast interpolated path between the open and closed conformations.
- Due to differences in residue counts, an intermediate closed-state structure was extracted and minimized for better alignment.
- The trajectory was recomputed starting from this refined state.
- P-NEB Module was used to optimize the path further, making it more physically accurate by relaxing the interpolated frames.
The processing steps were completed in minutes on a standard laptop, demonstrating that complex conformational simulations are accessible even without high-end computational resources.

Post-processing with P-NEB optimized the motion path.
Why This Matters
A common challenge for molecular modelers is bridging static structural data with dynamic behavior. Experimental structures show snapshots — but viruses function in motion.
By animating experimentally known conformations into a dynamic trajectory, the SAMSON approach allows researchers to:
- Visualize potential binding opportunities across the conformational path.
- Extract intermediate structures for docking simulations.
- Gain intuition on how conformational states relate to infectivity or antibody neutralization.
Researchers can download the PDB trajectories or SAMSON format file to explore or use as starting points for more detailed molecular dynamics or docking studies. It’s a valuable resource for those working in COVID-19 research or protein conformation analysis in general.
To learn more and access the simulation files, visit the original documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON here.