





When working with protein structures in molecular modeling, small inconsistencies can turn into major computation roadblocks. If you’re setting up a conformational transition, umbrella sampling, or just trying to interpolate between two protein forms, issues like alternate atom locations, water molecules, or small disconnected fragments often cause errors—or worse, misleading results.
One common error users encounter in SAMSON’s ARAP Interpolator is:
“Cannot proceed because the structure does not make one connected component”
This typically means the input structure contains parts such as water, ions, alternate location residues, or ligands, which break the continuity required for ARAP to function properly. Fortunately, avoiding this issue is straightforward with SAMSON’s integrated cleaning workflow.
Cleaning Up Your Structures
To get your proteins ready for interpolation—or any type of structural analysis—you should:
- Remove alternate residue locations (multiple positions for the same atom in PDB files).
- Eliminate solvent molecules like water that are irrelevant for interpolation paths.
- Delete ligands or ions unless they are critical to the conformational transition being modeled.
These steps ensure that the input conformations are connected molecular graphs—an absolute requirement for ARAP interpolation to work properly.
One-Click Protein Preparation in SAMSON
SAMSON makes structure preparation easy via the Home > Prepare functionality:
- Load your structure (or fetch it directly from the PDB using
Home > Fetch). - Go to Home > Prepare and select the structure to be cleaned.
- The algorithm will clean up the structure, removing non-relevant parts automatically.
This step only takes a few seconds, but it saves you a lot of debugging time later. Clean structures also yield smoother, more biologically realistic interpolation paths.
Why It Matters
Structure continuity is essential for building a reliable As-Rigid-As-Possible (ARAP) geometric model. ARAP treats molecular structures as elastic networks; if your structure is disconnected, the algorithm doesn’t know how to deform parts of the molecule in a coordinated way.
Moreover, intermediate frames resulting from interpolation will not be meaningful if extraneous fragments move independently of the protein’s backbone. This could affect downstream workflows, including umbrella sampling setups, MD simulations, or conformational animations.
Real Case: Diphtheria Toxin Conformations
A great example comes from the tutorial that interpolates between two conformations of the Diphtheria Toxin. The original 1MDT structure includes two chains (A and B), but the transition path only concerns chain A. Deleting chain B and cleaning up the structure ensures ARAP can generate a smooth interpolation path between 1DDT A and 1MDT A.
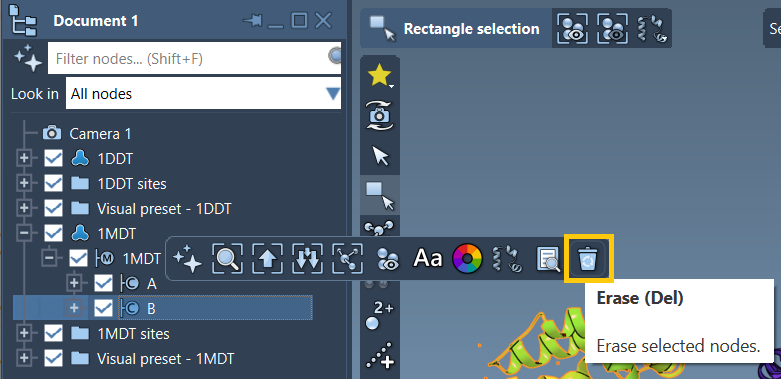
This step might seem minor, but it is critical for successful path computation.
Takeaway
Clean structures are not just good practice—they’re essential for geometric interpolation and downstream simulations. If you’ve ever run into mysterious ARAP errors or poor interpolation results, structure preparation is the first thing to check.
Explore the full tutorial for protein interpolation here: ARAP Interpolation for Protein Structures
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.