





When you’re trying to optimize the properties of a lead compound, exploring small chemical changes at specific sites can be a powerful strategy. Medicinal chemists often want to tweak one part of a molecule and see how it affects activity or binding. But doing that manually—especially across multiple positions in a complex structure—can become time-consuming and error-prone.
This is where positional analogue scanning becomes useful. And thanks to the SMILES Manager extension in SAMSON, you can now apply this strategy interactively without writing scripts or switching between multiple tools.
What is positional analogue scanning?
It’s a method that systematically introduces modifications—like changing a carbon atom to nitrogen or adding small groups—at defined positions within a molecular structure. The idea is to evaluate how each specific substitution potentially alters molecular properties, such as activity, selectivity, or binding affinity.
This approach has been discussed in detail in a 2019 paper by Pennington and Muegge as well as on Pat Walters’ cheminformatics blog. In SAMSON, the method is now accessible in a few interactive steps, making analogue generation significantly more convenient for modelers.
Step-by-step: Scan your molecule with SMILES Manager
In the SMILES Manager, the process begins with selecting a single molecule—either from your workspace or by entering its SMILES code. Then, you define a pattern to scan for. In the example tutorial, aromatic hydrogen atoms are targeted using the SMARTS pattern [cH].
Once the pattern is recognized, you’re given options to:
- Replace the atom(s), e.g., change carbon to nitrogen;
- Attach small groups, like fluorine or methyl.
Then, simply click Run and the tool will automatically generate a list of molecular variants along with their corresponding SMILES strings and 2D depictions.
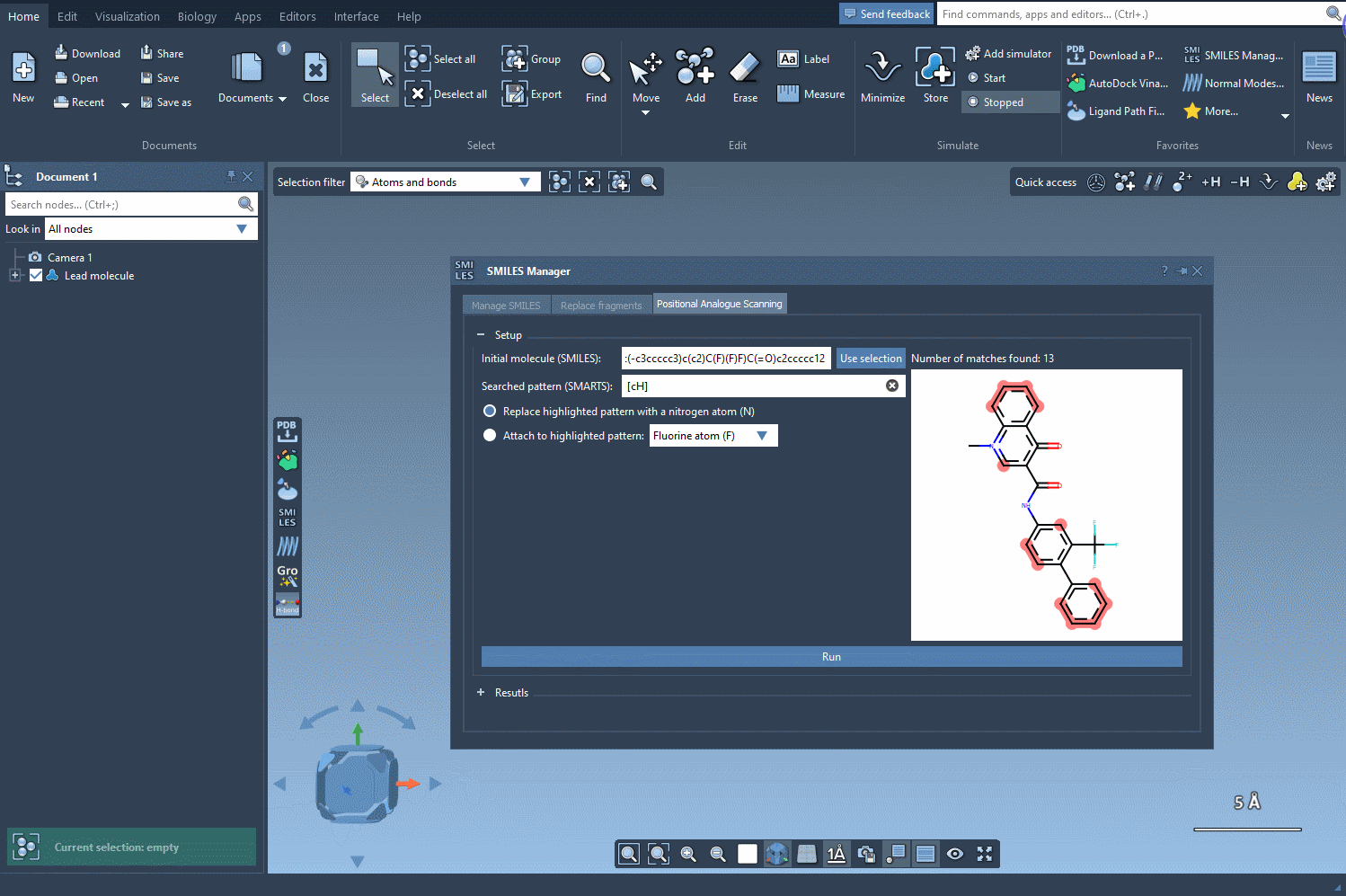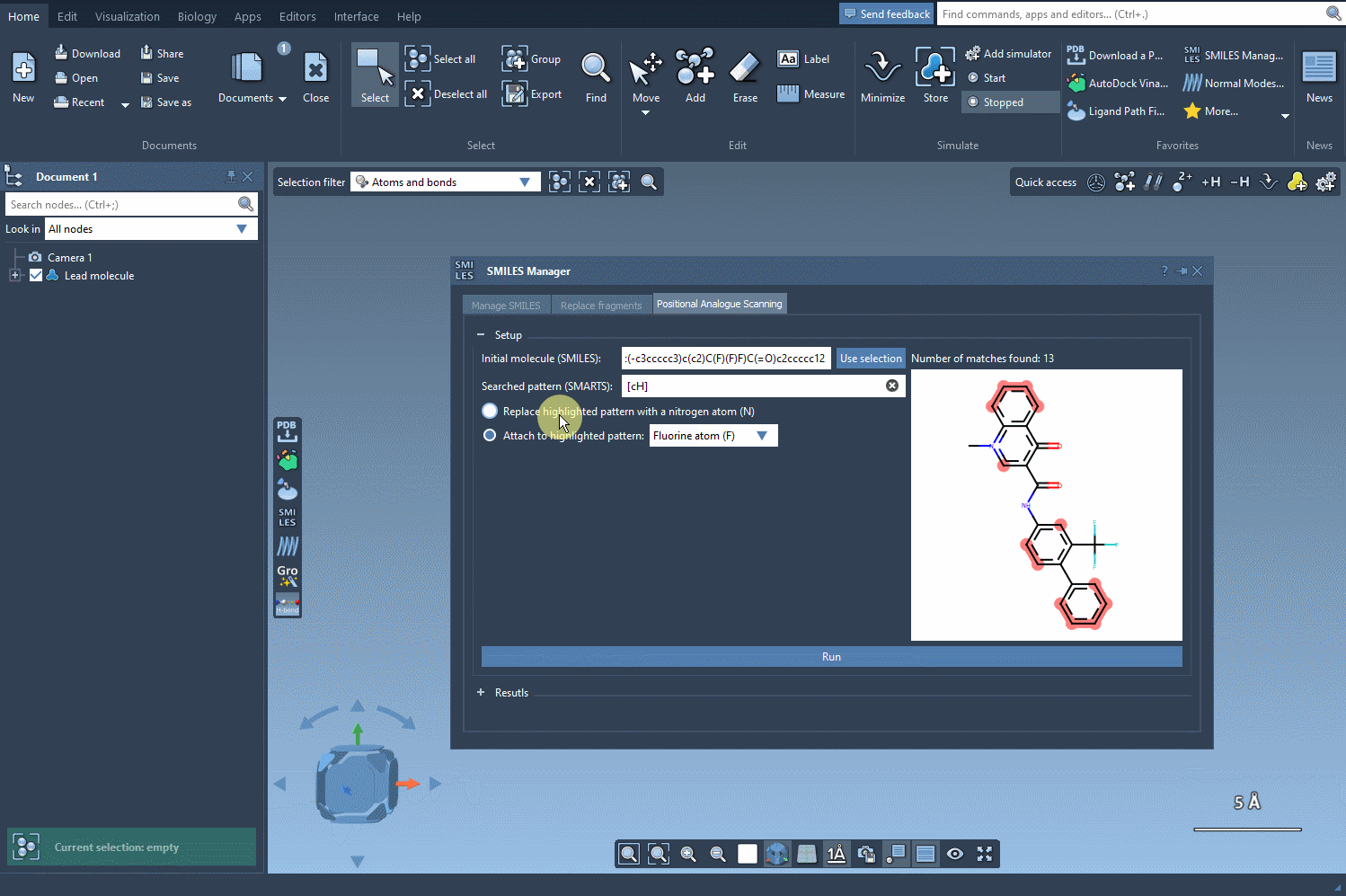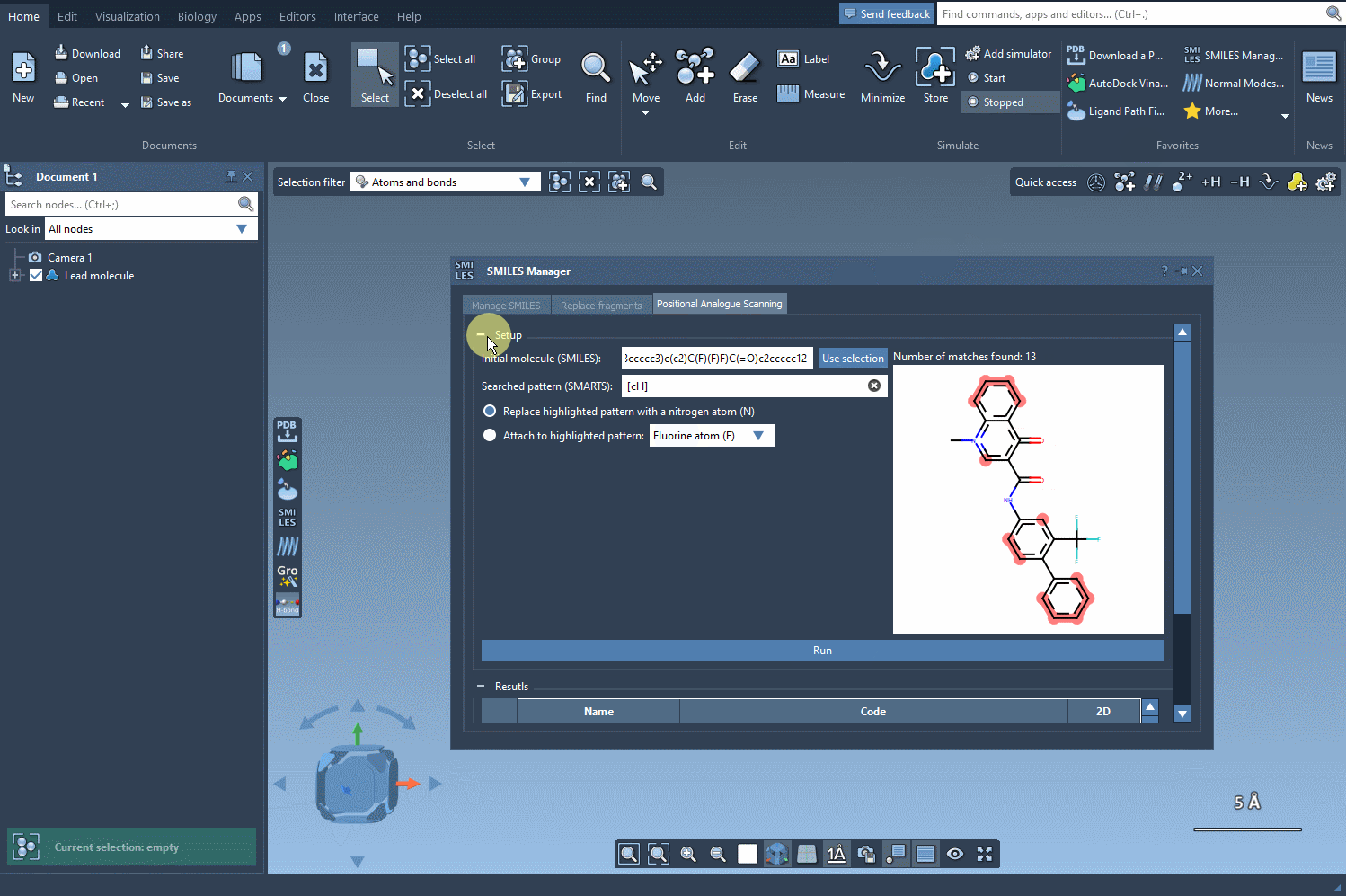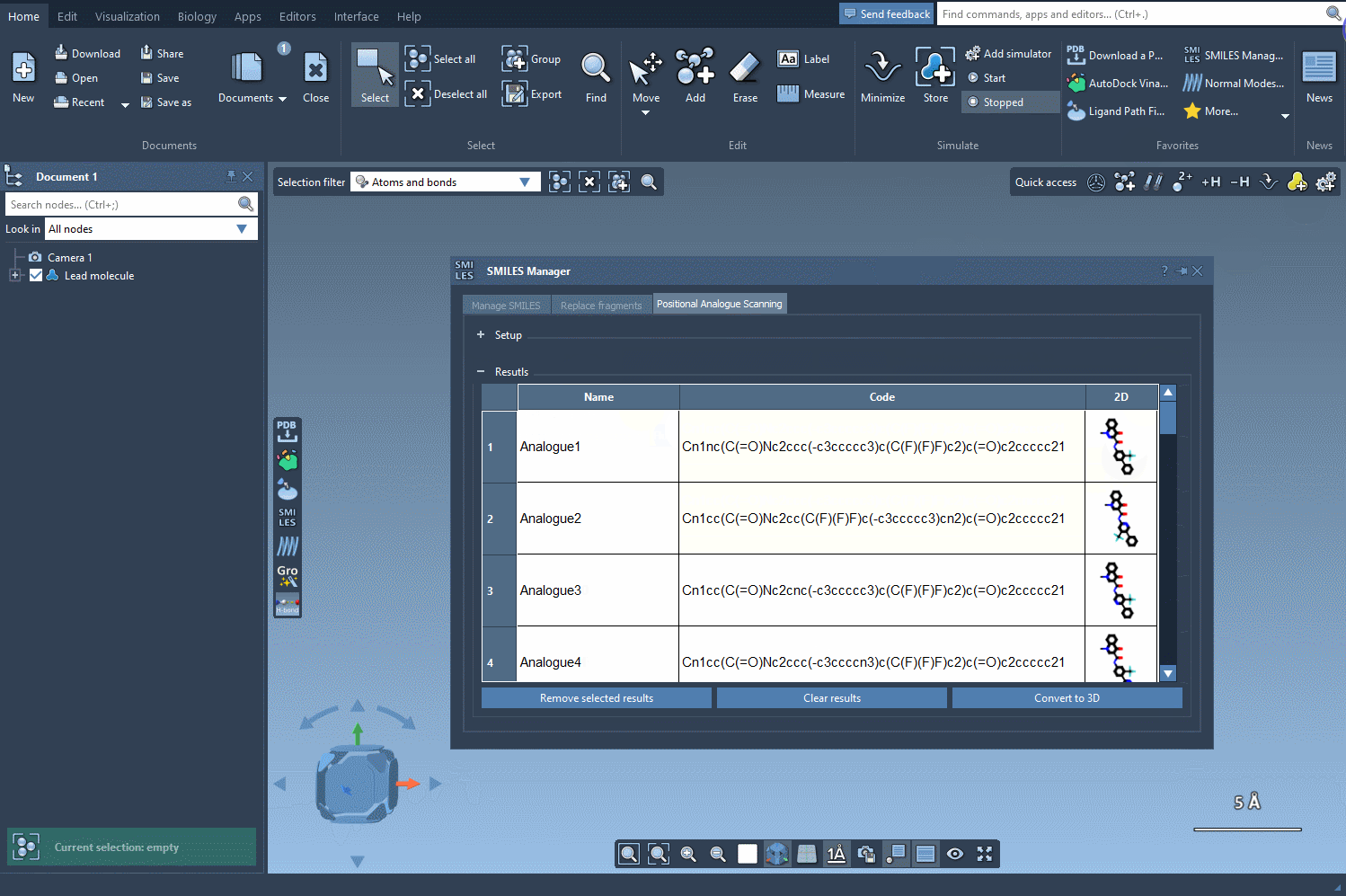
This interface allows you to visually inspect each analogue and even edit names or SMILES codes directly from the table. More importantly, you can convert any analogue into a full 3D molecular structure for further study or simulations.
Why it matters
This feature solves a common bottleneck in early-stage drug design or molecular property optimization: the lack of a rapid and flexible way to test variants. Instead of clicking through menus or writing syntax-heavy scripts in other platforms, SMILES Manager lets you do this work visually and interactively in SAMSON.
Whether you’re designing inhibitors, probes, or just exploring structure–activity relationships, this workflow speeds up early-stage iterations tremendously. It’s a time-saver, especially during brainstorming or hit-expansion phases, and can tie directly into downstream workflows like docking or molecular property analysis.
Learn more
The full tutorial with more details and examples is available here.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at www.samson-connect.net.