





For molecular modelers studying viral pathogens, understanding conformational changes in key proteins is more than just a matter of curiosity—it’s essential for tasks like drug design, antibody targeting, and vaccine development. One significant structure that’s drawn global focus is the SARS-CoV-2 spike protein. A question that often arises is: how exactly does the spike move between its closed and open states, and how can we study that motion computationally?
This post outlines how the conformational motion of the SARS-CoV-2 spike protein was computed using SAMSON, offering detailed visuals and downloadable data to help you start or enhance your own computational explorations.
The Spike Protein in Motion
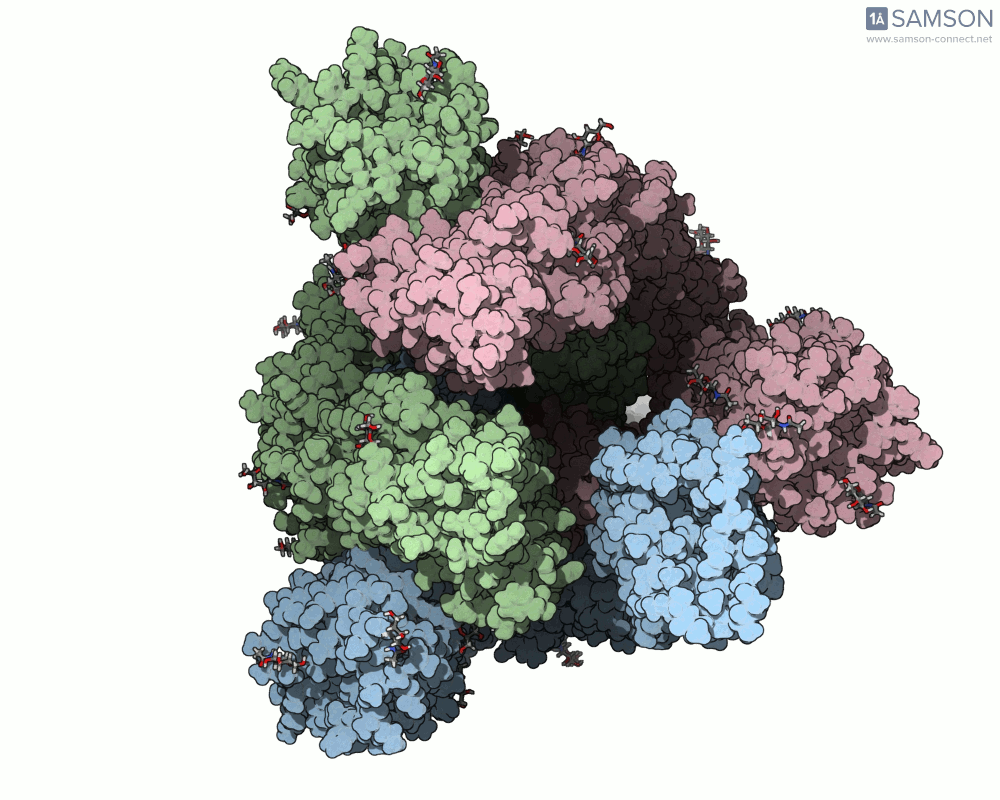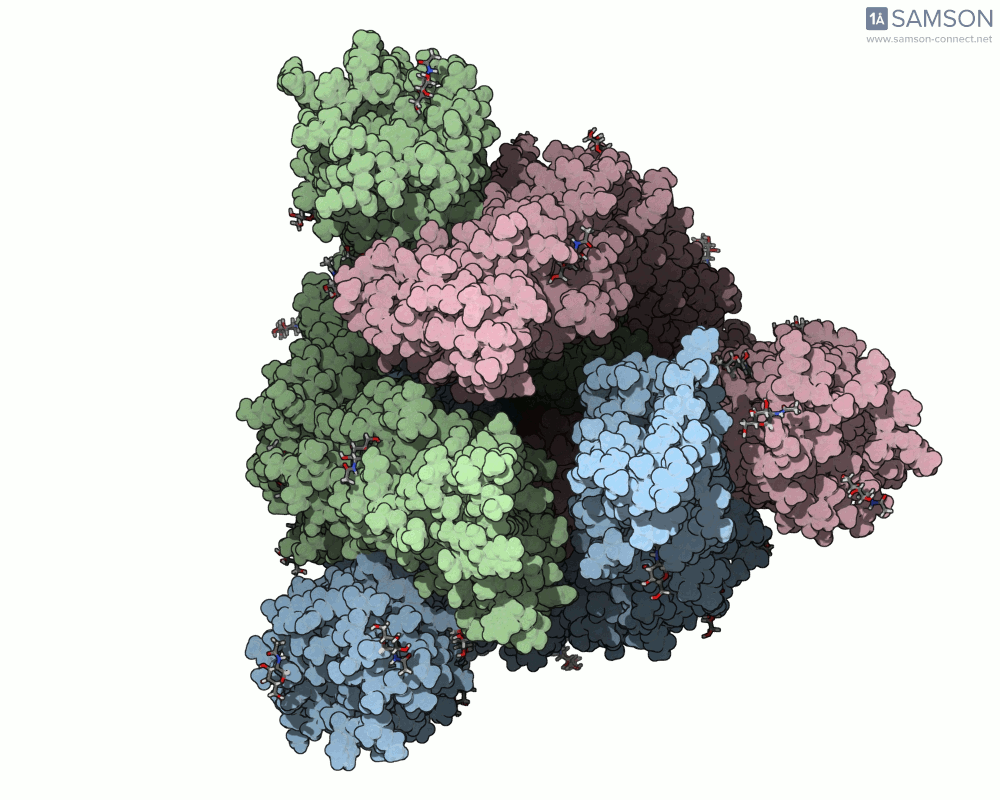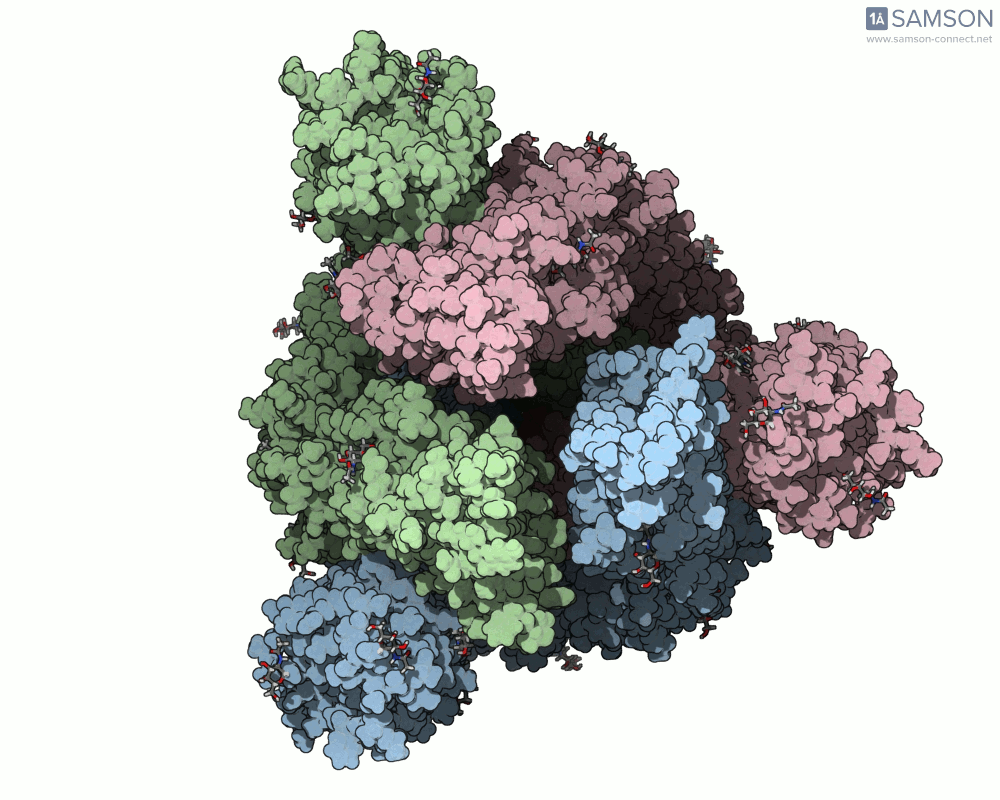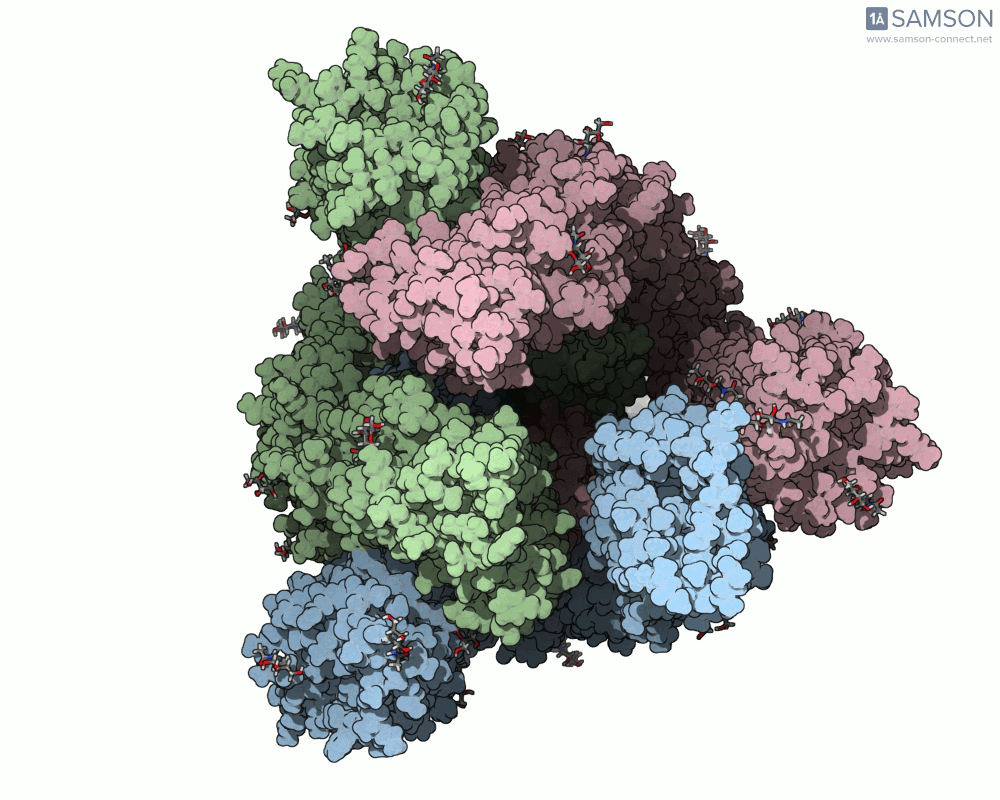
The motion of the SARS-CoV-2 spike from its closed (non-binding) state to its open (ACE2-binding) state was simulated using a combination of interpolation and optimization approaches. The transformation is key to viral entry into host cells, as binding with the ACE2 receptor only occurs when one part of the spike opens.
Here’s what that motion looks like from different angles:
Side view:
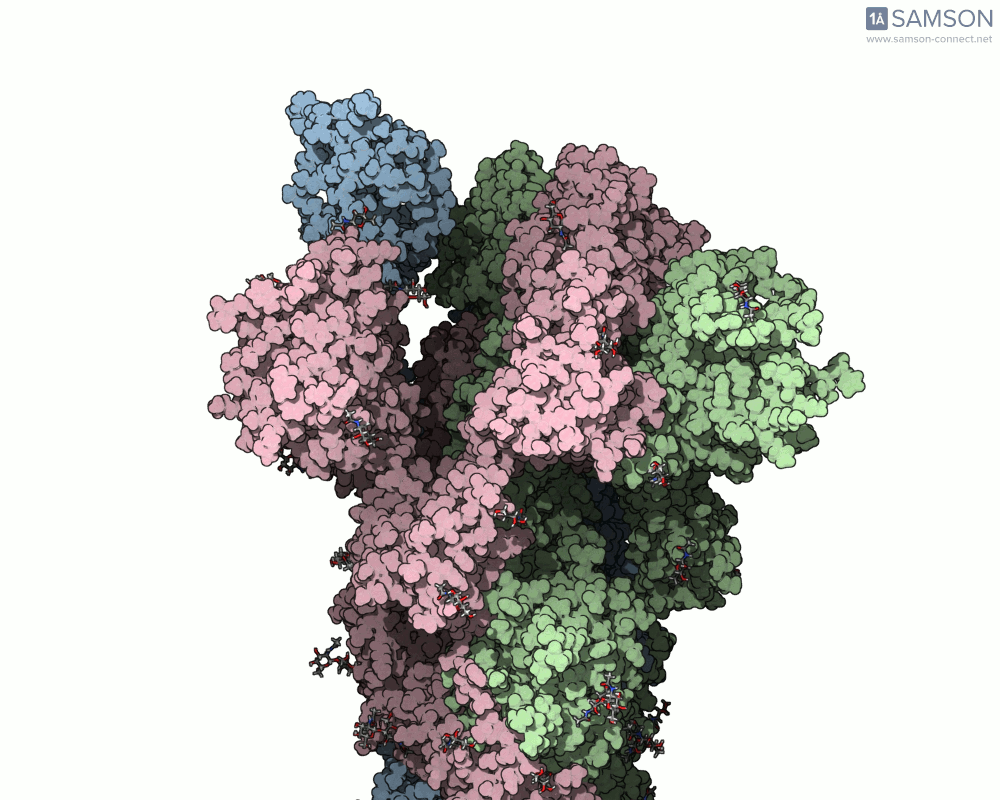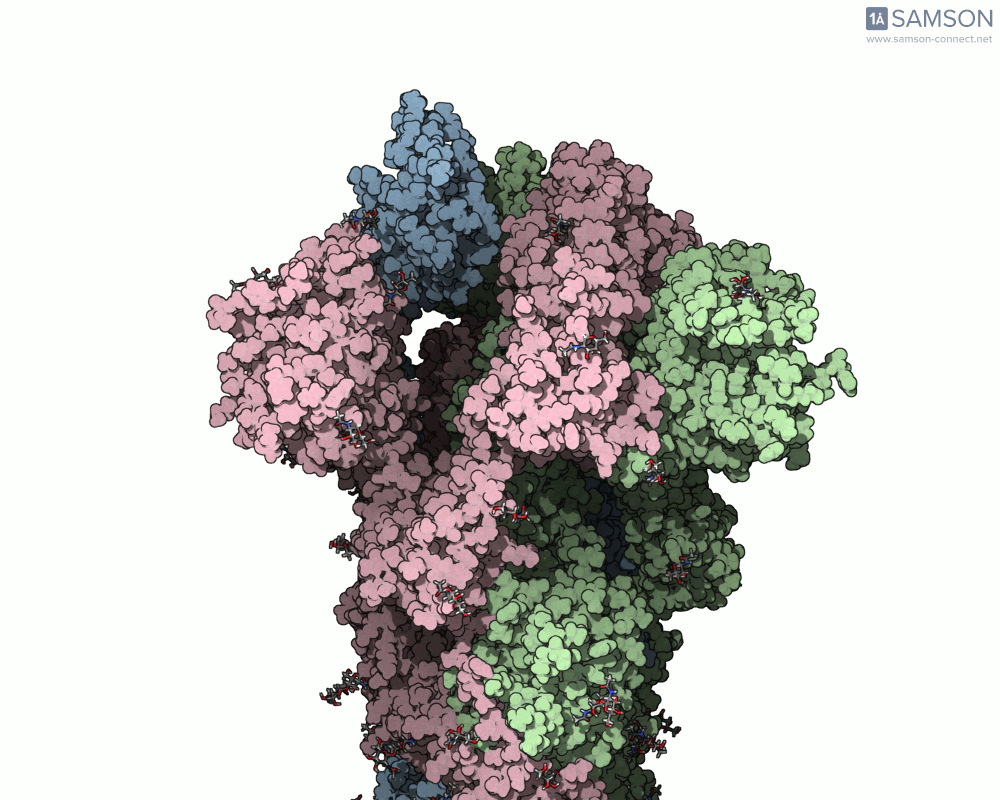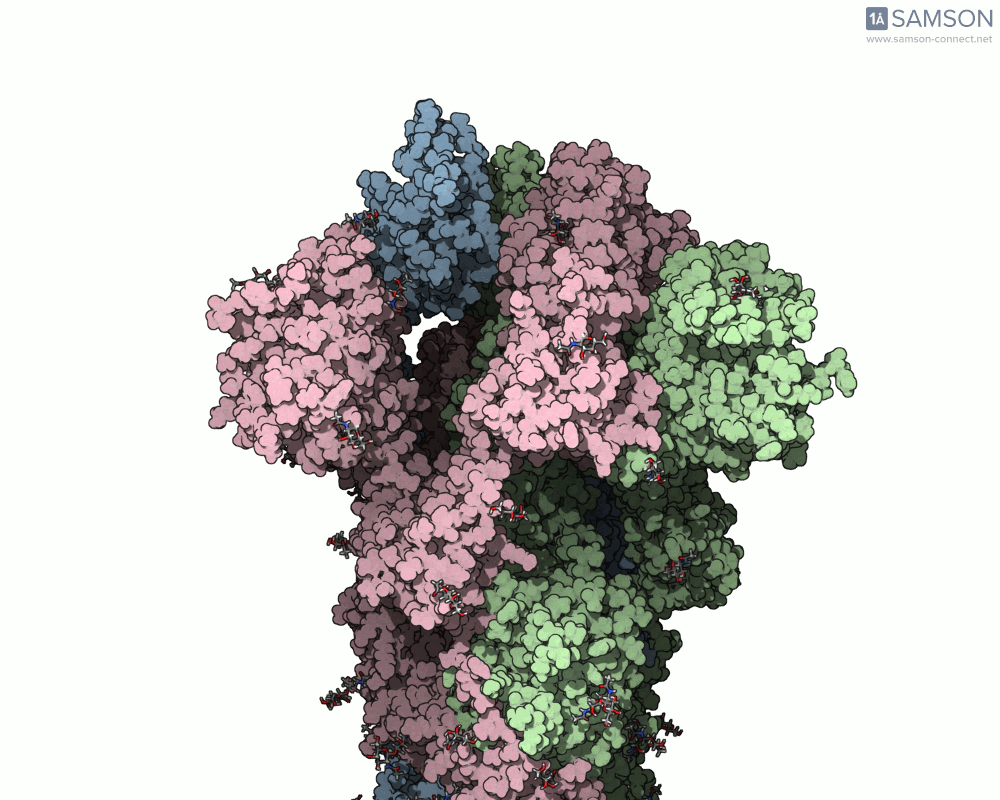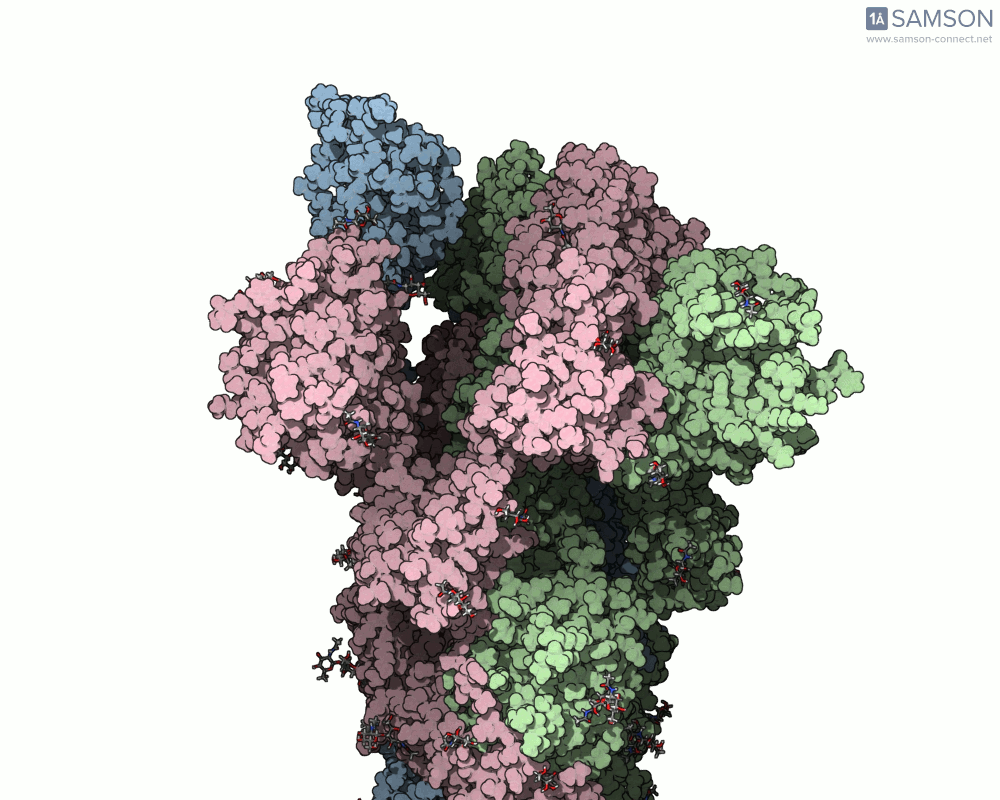
Another angle:
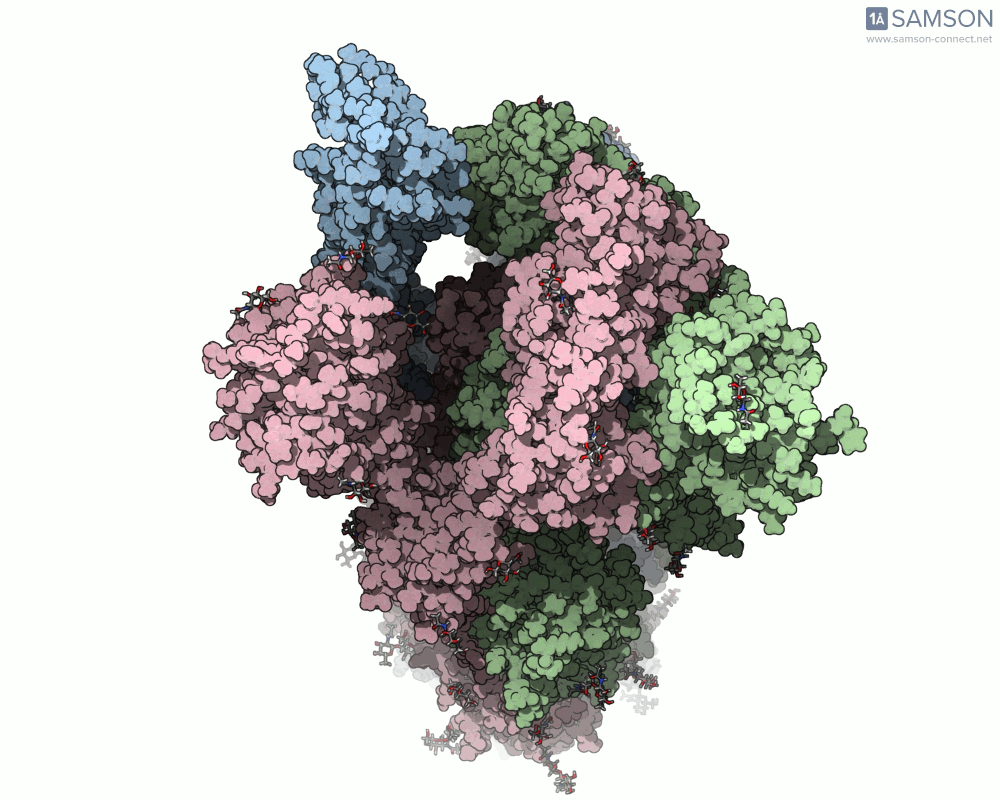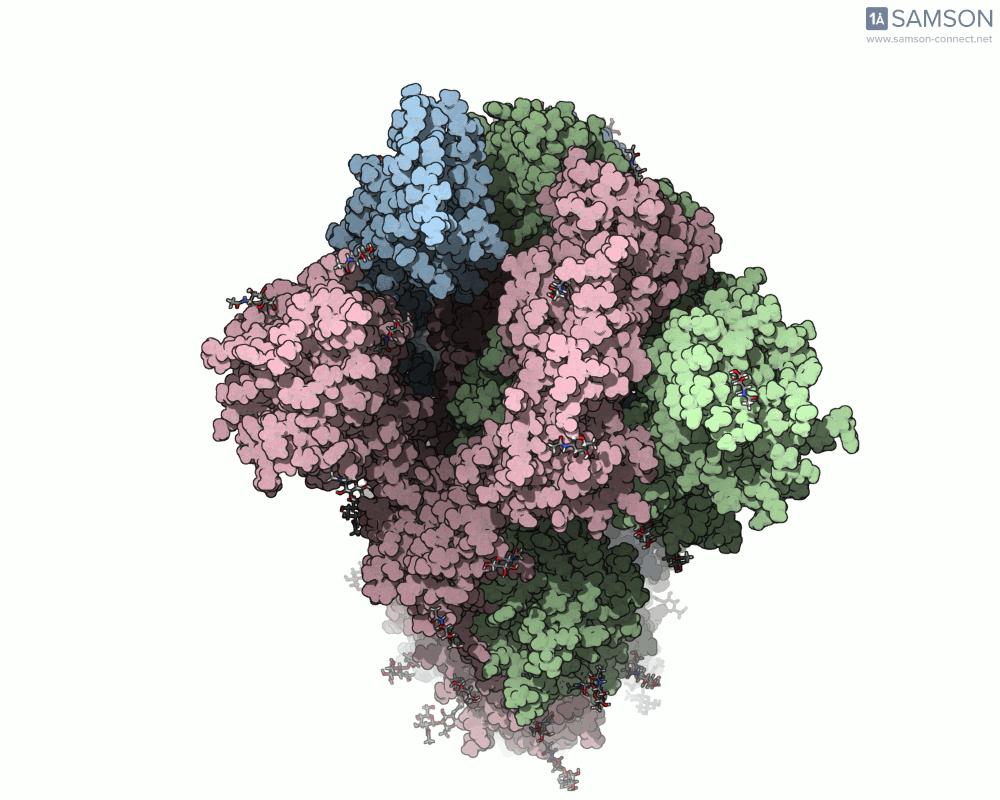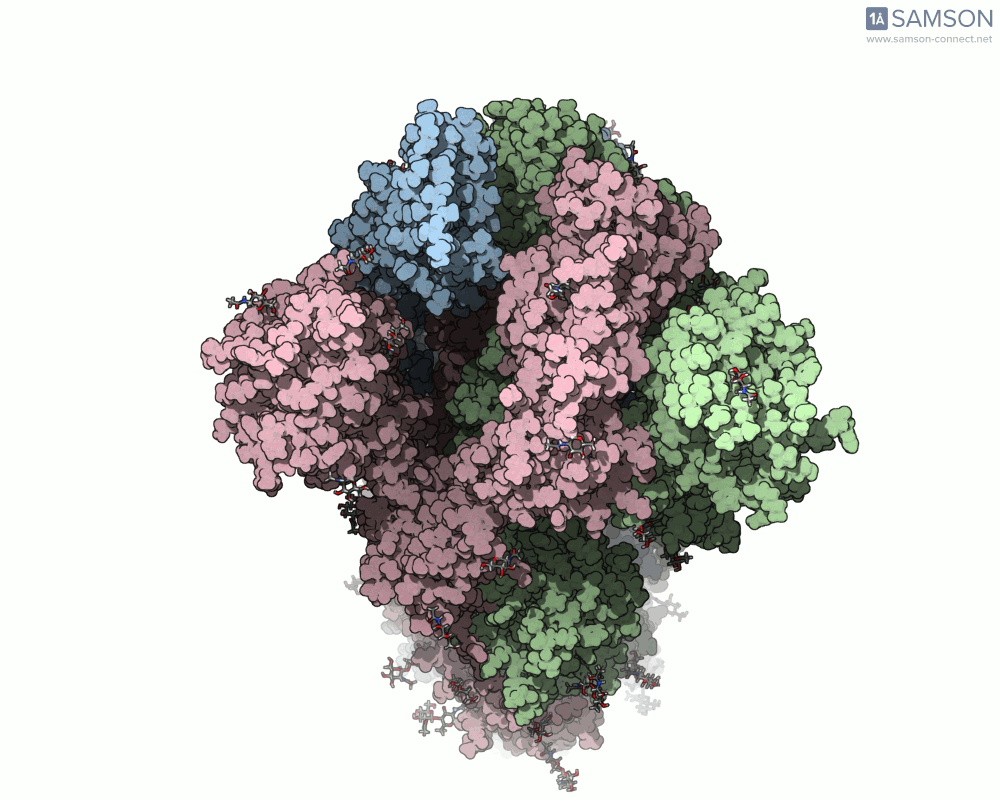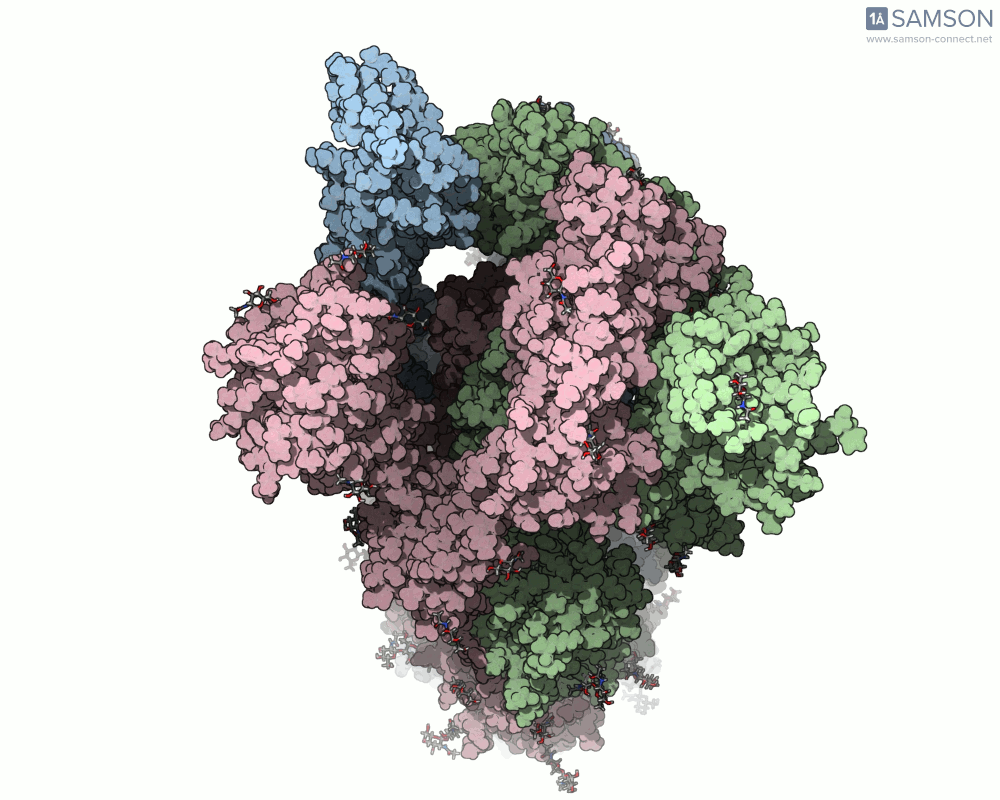
Top view:
Computed Trajectories Available
You can directly download the computed trajectories in multiple formats. These serve as a foundation for your own simulations or visual explorations:
- Set of PDB trajectory files
- Single PDB file with the trajectory
- SAMSON file with interactive trajectory
The SAMSON file is particularly helpful, containing both the open and closed conformations, as well as path data prepared with two tools: the As-Rigid-As-Possible (ARAP) Interpolation Path module and the Parallel Nudged Elastic Band (P-NEB) module.

How Was the Motion Computed?
The trajectory was computed starting from two crystallographic structures of the spike protein:
These were pre-processed to harmonize sugar structures and hydrogen atoms, followed by interpolation using the ARAP module. A subsequent refinement was applied using the P-NEB module.
This workflow results in a realistic and smooth conformational transition—achieved in under 20 minutes on a standard laptop—that can be visually inspected and exported for further analysis or molecular docking simulations.
Whether you’re designing inhibitors, modeling antibody binding, or just exploring structural virology, having access to a clean, manipulable trajectory like this can speed up insight generation and hypothesis testing.
Want to try it yourself? Explore the full documentation here.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.