





Many molecular modelers face the repetitive task of exploring small chemical modifications to a lead compound. It’s increasingly common in early-stage drug discovery to generate large libraries of analogues by substituting small fragments, in order to evaluate changes in properties, binding, or stability. But doing this manually? Tedious, repetitive, and prone to error.
The SMILES Manager extension of SAMSON offers a feature specifically designed to streamline this task: Replace fragments. If you’ve ever wanted to run quick bioisosteric replacements, or investigate chemical variations without spending days on structure editing, this is a tool worth knowing about.
Why Does Fragment Replacement Matter?
Fragment replacement lets you explore chemical space efficiently by swapping out part of a molecule—such as a functional group or linker—with alternatives. For example, replacing an amide bond with a sulfonamide or a triazole might help fine-tune pharmacokinetics or avoid metabolic liabilities. But doing this manually in a modeling environment often requires drawing the same molecule over and over—just for a single substitution. That’s time you could be spending on analysis.
How the “Replace fragments” Tab Works in SMILES Manager
In the Replace fragments tab, the process is intuitive and fast:
- Import an initial molecule – Either paste a SMILES or load from a file. For example:
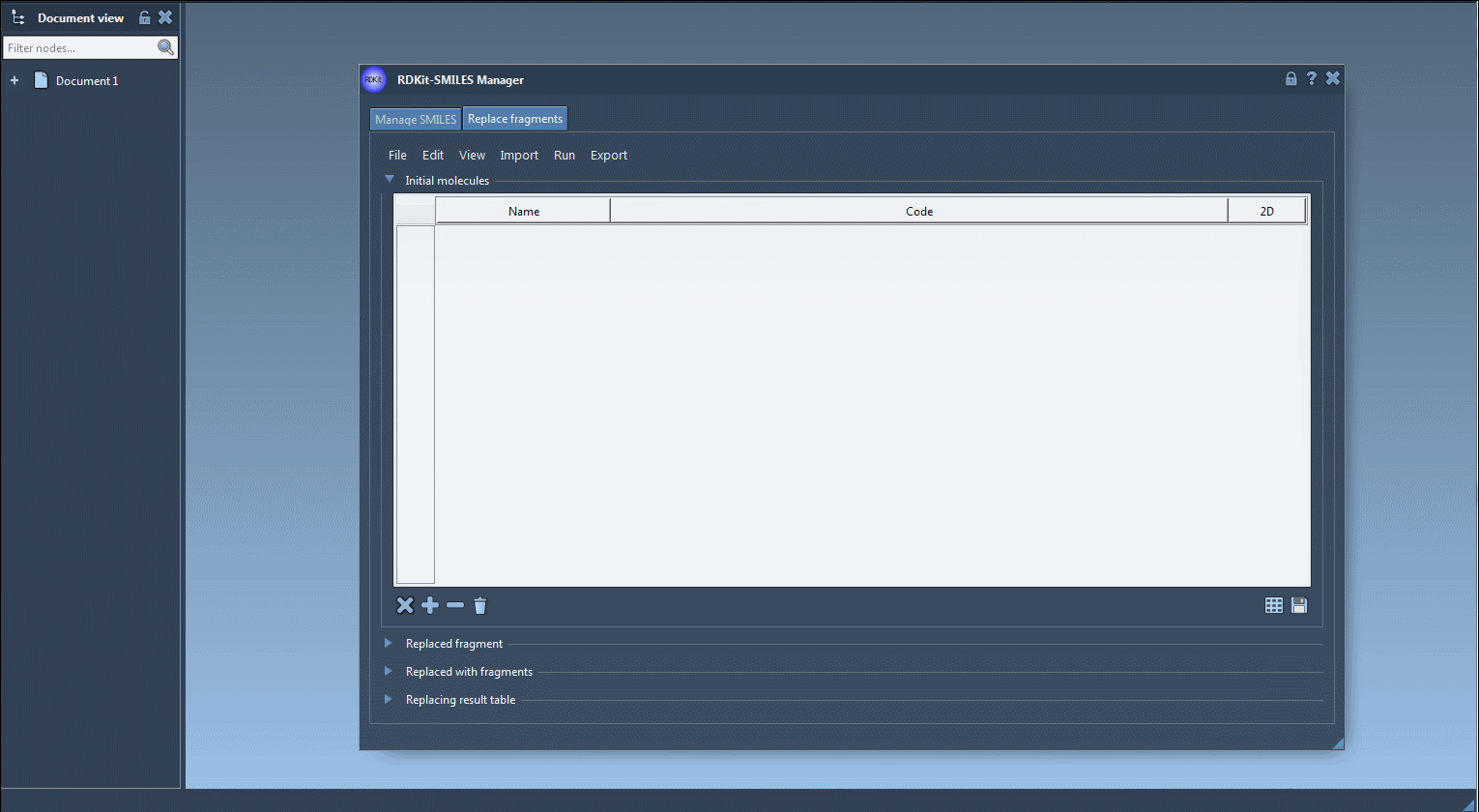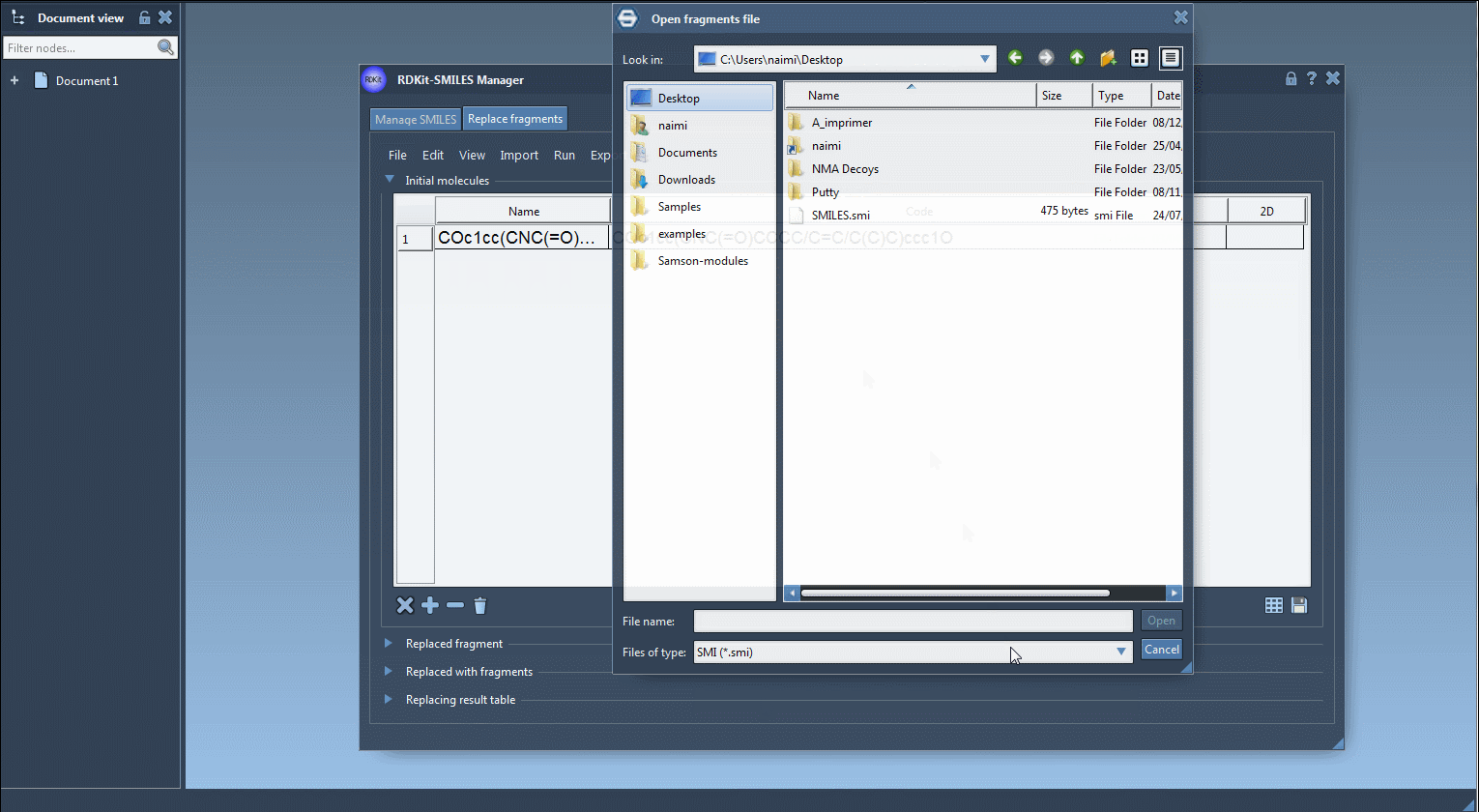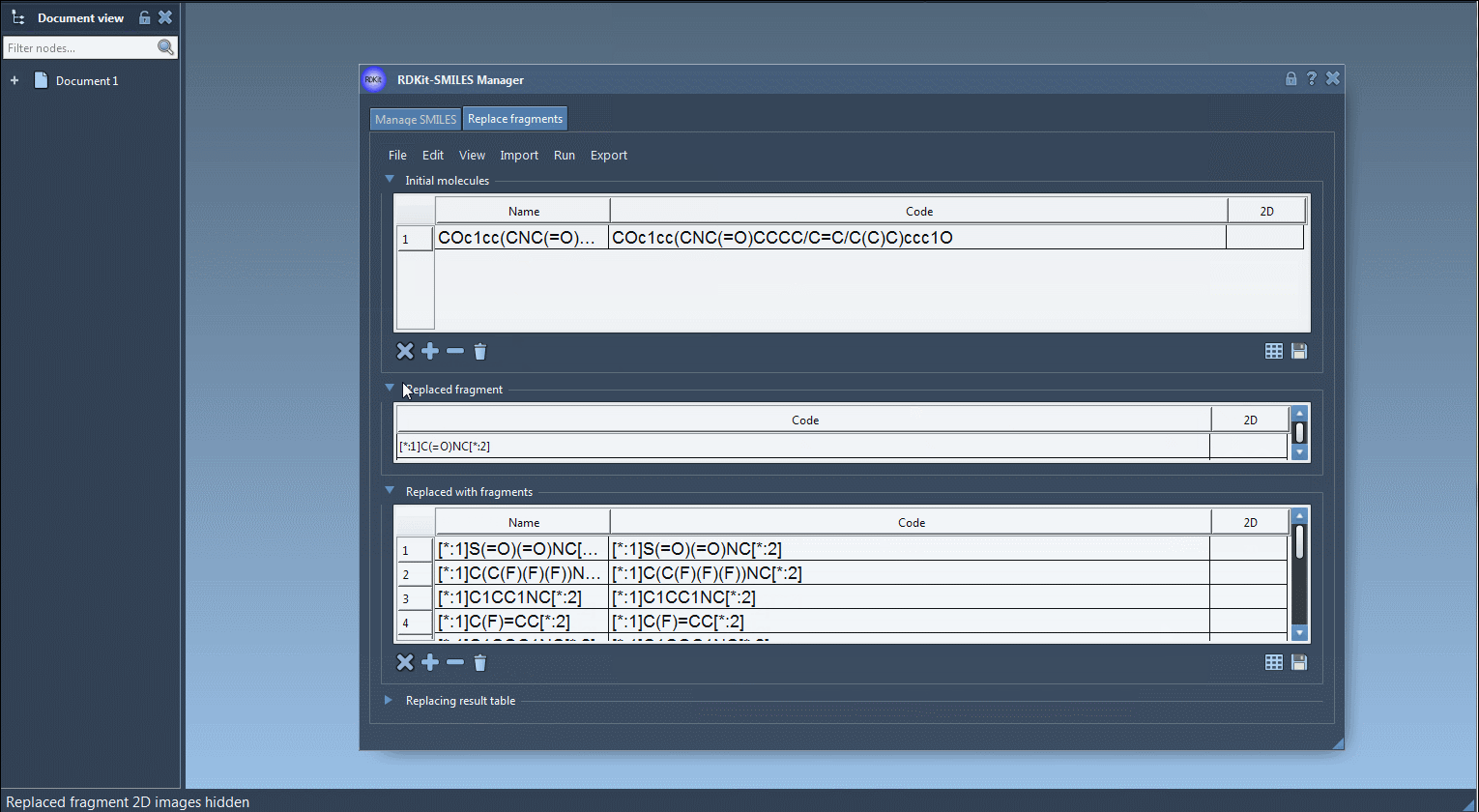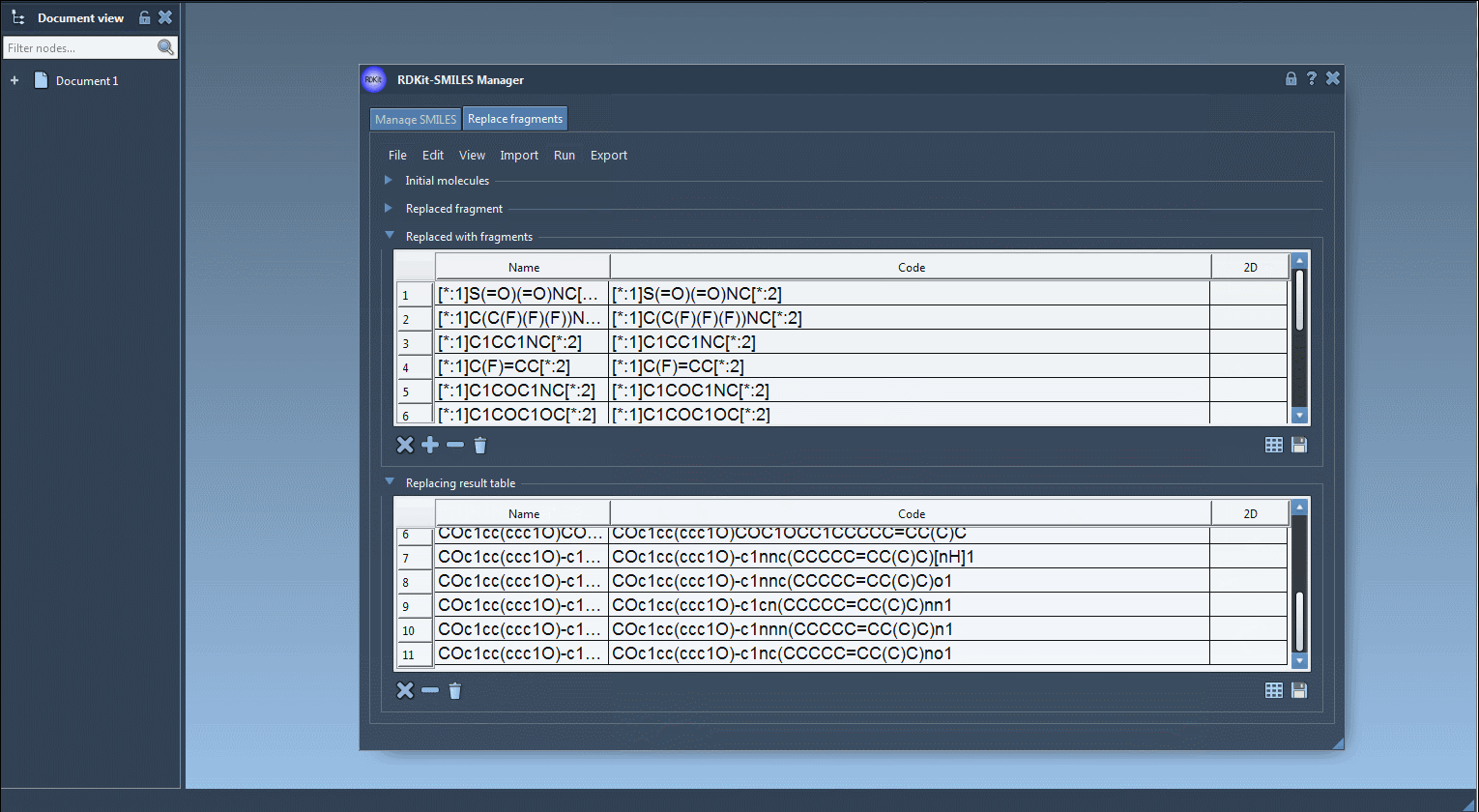
COc1cc(CNC(=O)CCCC/C=C/C(C)C)ccc1O - Define the fragment to replace – Specify the substructure using SMARTS notation. For example:
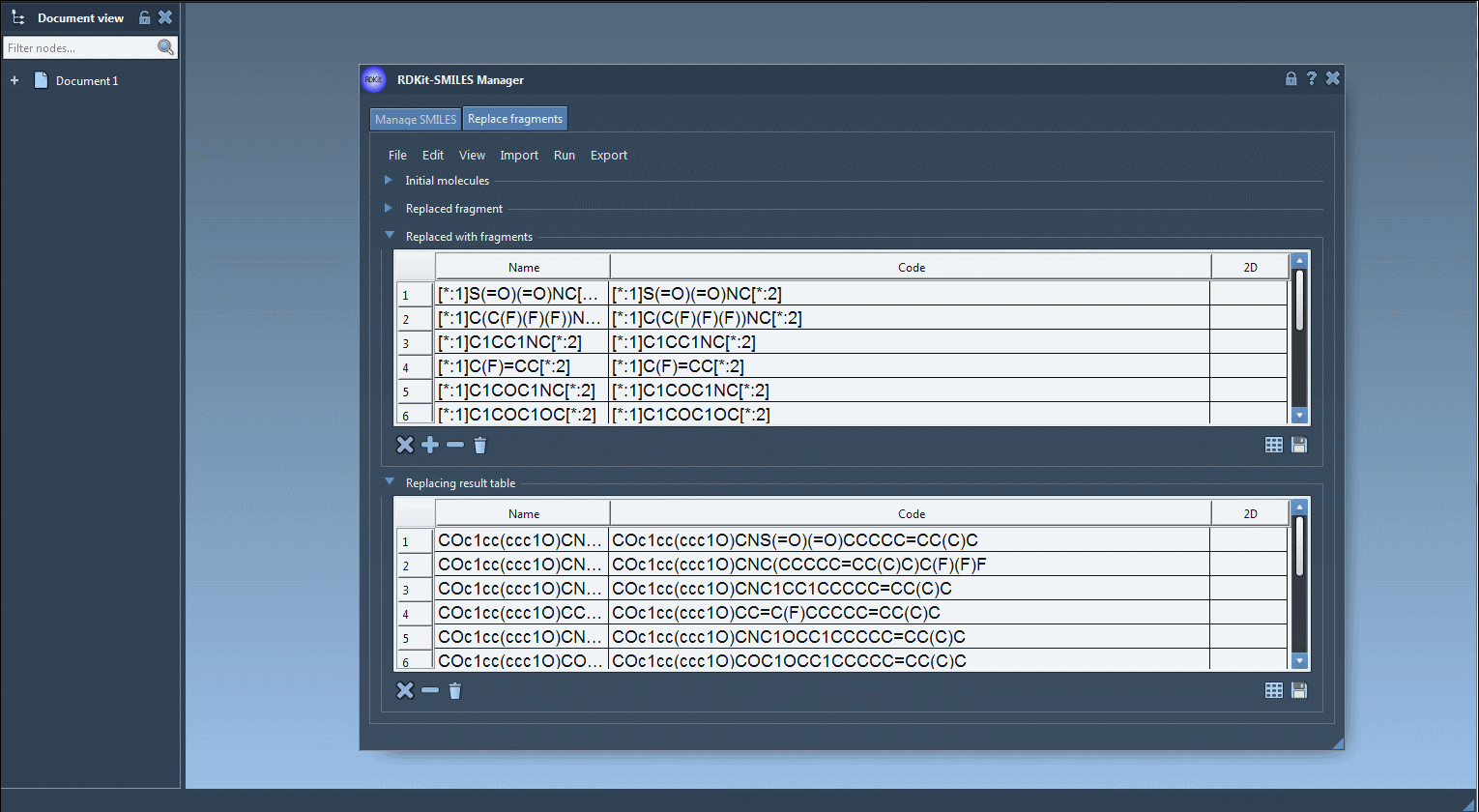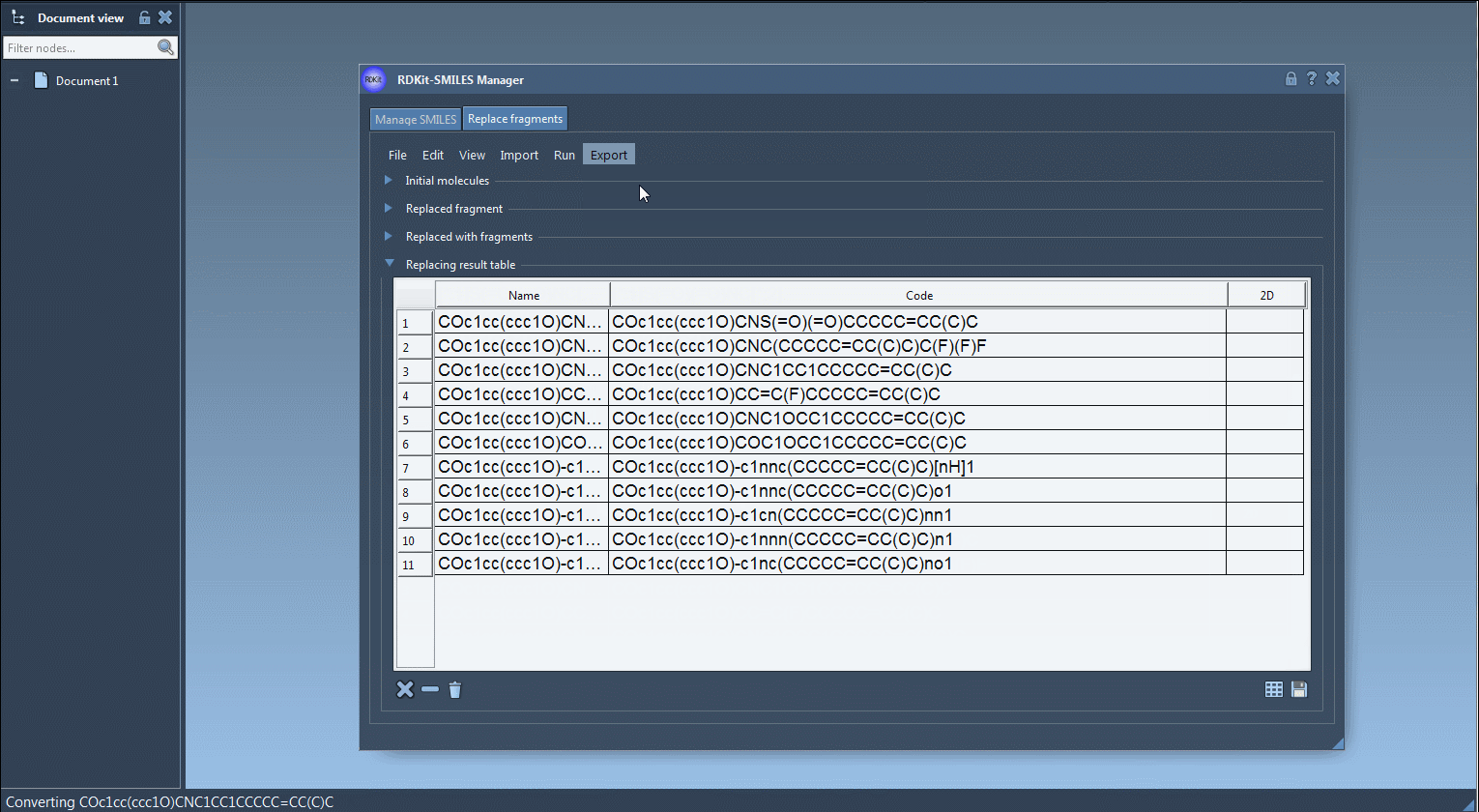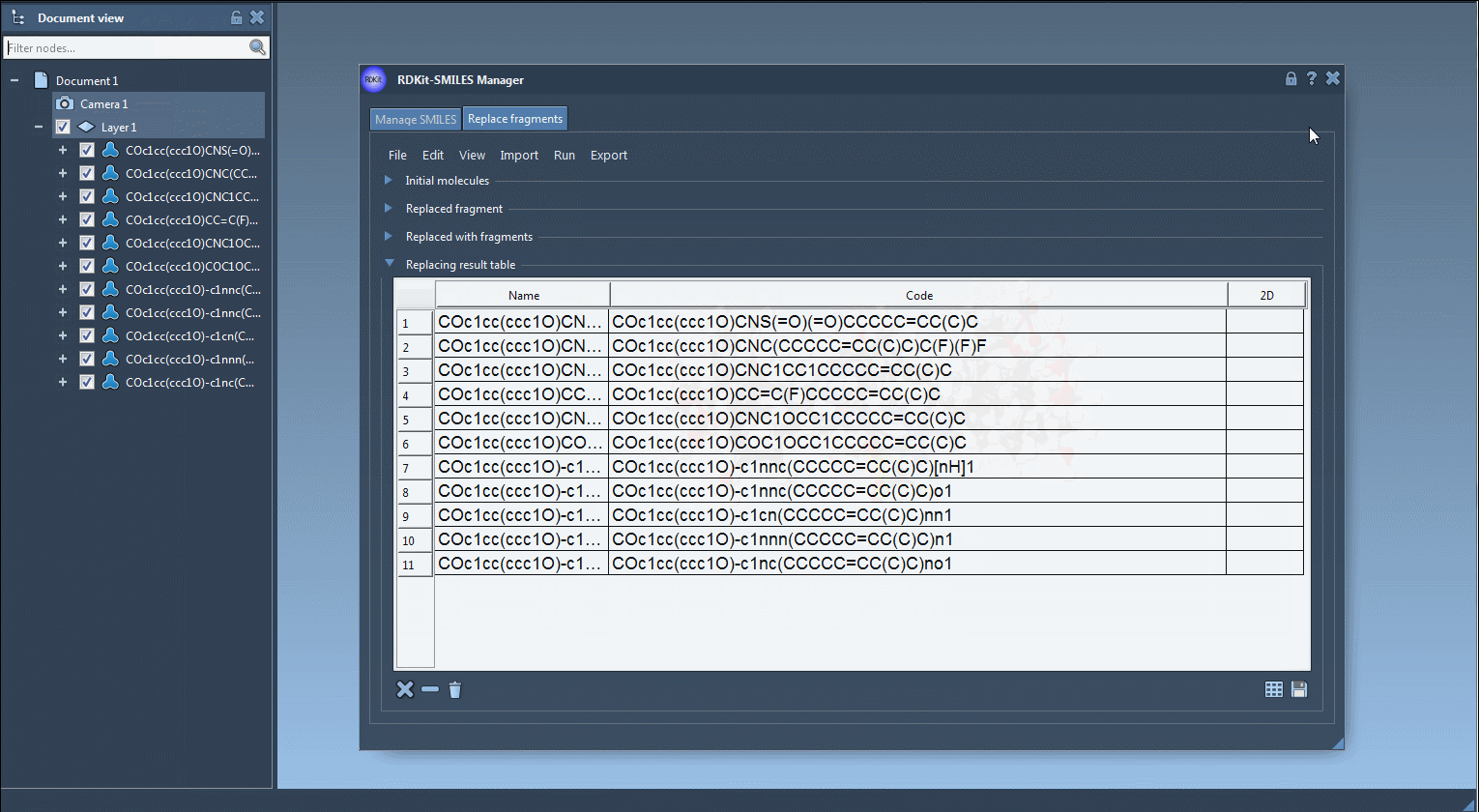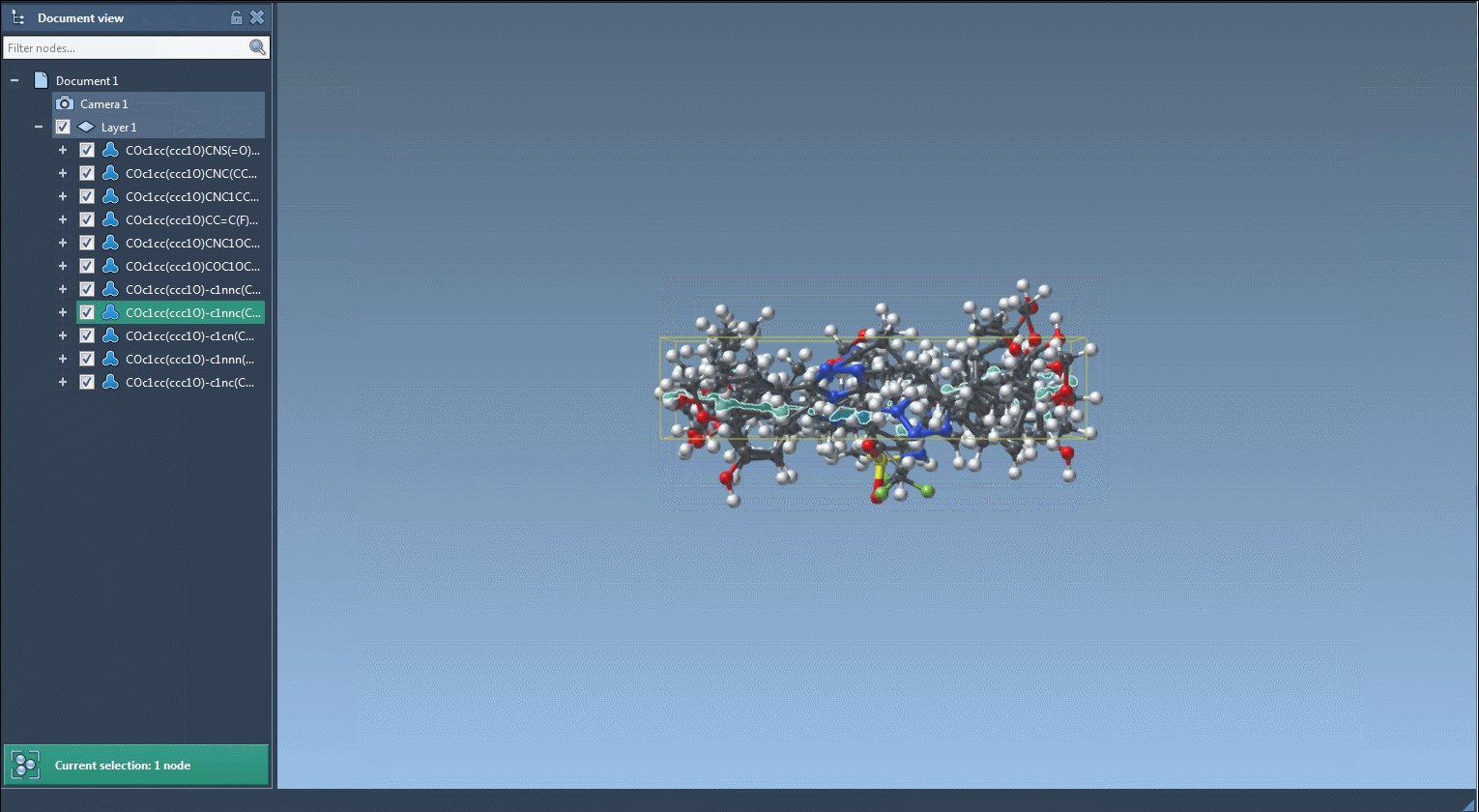
[[*:1]C(=O)NC[*:2]] - Provide a list of replacement fragments – These could be anything from sulfonamides to rings or heterocycles:
[*:1]S(=O)(=O)NC[*:2],
[*:1]C(C(F)(F)(F))NC[*:2],
[*:1]C1COC1NC[*:2],
...
- Click “Replace Fragments” in the Run menu – The SMILES Manager generates a full set of new molecules using the replacement rules above.
Then What?
Generated molecules are listed in a new results table. From here, they can be immediately exported as 3D structures into your active SAMSON document, saved, or visualized as a grid of 2D depictions.
If your goal is to create a diverse and targeted molecular library in minutes, this feature can make a big difference. No need to repeatedly draw molecules or write scripts. Everything is controlled in a couple of clicks and menu options. Every fragment inserted preserves proper valence rules and bonding through RDKit integration, ensuring chemically valid results.
Who Is This For?
This is useful for medicinal chemists, computational chemists, and educators working in molecular design, cheminformatics, or structural bioinformatics. If you use SMILES strings and routinely explore variations of lead compounds, this tool can enhance your workflows.
The best part? No programming required. You only need to understand basic SMILES and SMARTS notation, and the interface takes care of the rest.
If you’re curious to dive deeper or want a step-by-step walkthrough, check out the full tutorial at SAMSON’s official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from samson-connect.net.