





For molecular modelers, a common pain point is the efficient setup and simulation of molecular systems. The Universal Force Field (UFF) offers a powerful interaction model for simulating molecular interactions. However, ensuring a smooth configuration can often be challenging. In this guide, we’ll walk you through leveraging SAMSON to effortlessly prepare your molecular systems for UFF simulations, saving you time and effort.
What is the Universal Force Field?
The Universal Force Field (UFF) is a versatile force field originally proposed by Rappe et al., designed to facilitate molecular simulations across the entire periodic table. In SAMSON, UFF includes an automatic molecular perception system that computes bonds, bond orders, and atom types, ensuring compatibility even with complex molecular systems.
Getting Started with UFF in SAMSON
Setting up UFF in SAMSON is straightforward and intuitive. Here is a step-by-step approach:
- Prepare a document with your molecular system.
- Add a simulator by navigating to
Edit > Simulate > Add simulator. For quicker access, use the shortcut:Ctrl + Shift + M(Windows) orCmd + Shift + M(Mac). - Select Universal Force Field from the list of interaction models.
- Select a state updater, such as FIRE, for your simulation.
- Click OK. The UFF setup window will appear.
Customizing UFF Setup
The UFF setup window allows you to tailor its configuration based on your simulation requirements:
- Use existing bonds: Check this option if pre-defined bonds exist in your model. Otherwise, UFF will automatically compute bonds based on atom types and positions.
- Press OK to initialize the UFF system. This triggers SAMSON to perform the necessary molecular structure perceptions, such as calculating covalent bonds, bond orders, and atom types.
Should there be inconsistencies, a detailed warning will appear, guiding you towards resolutions. Once initialized, the UFF parameter window provides further configuration options, including:
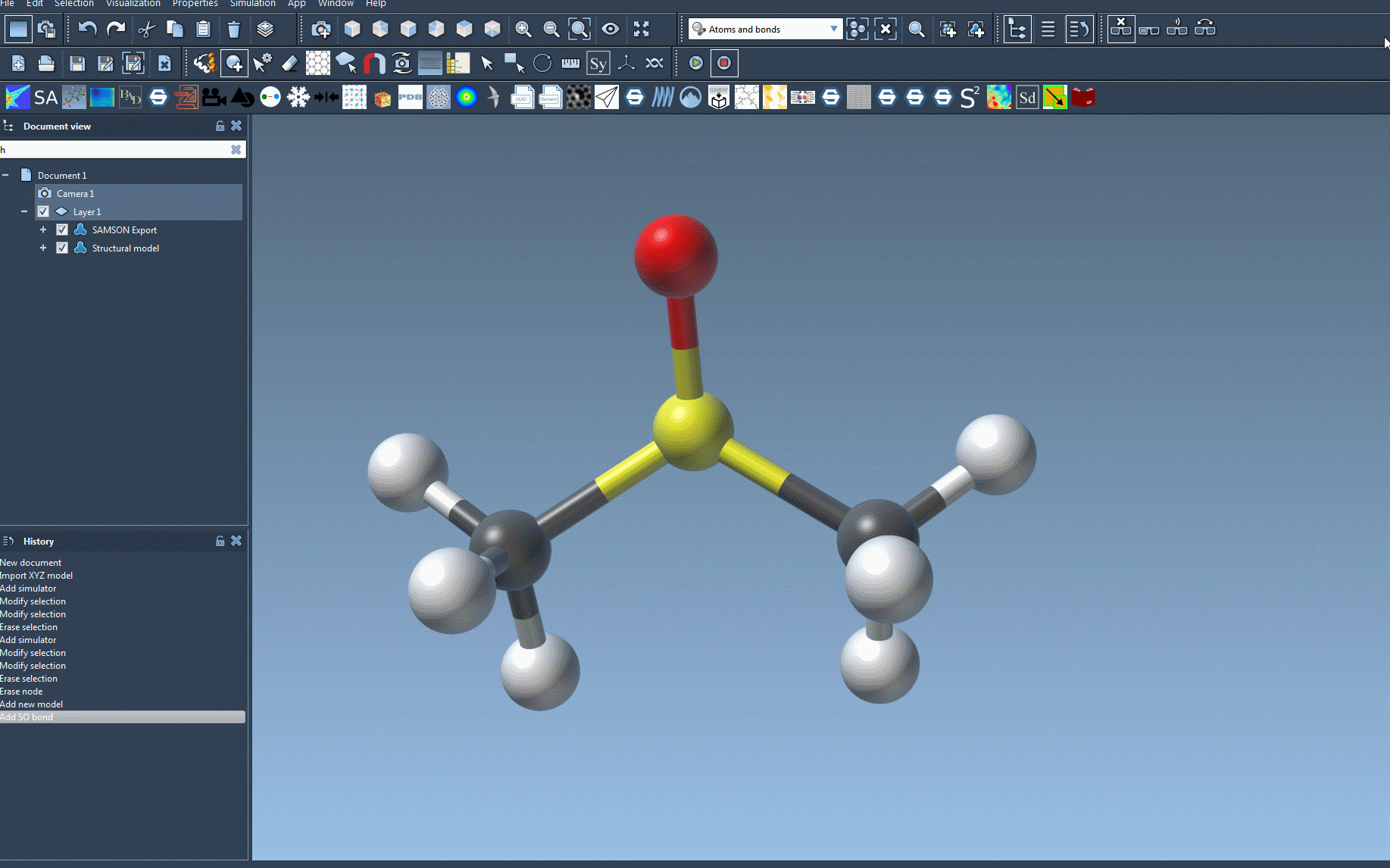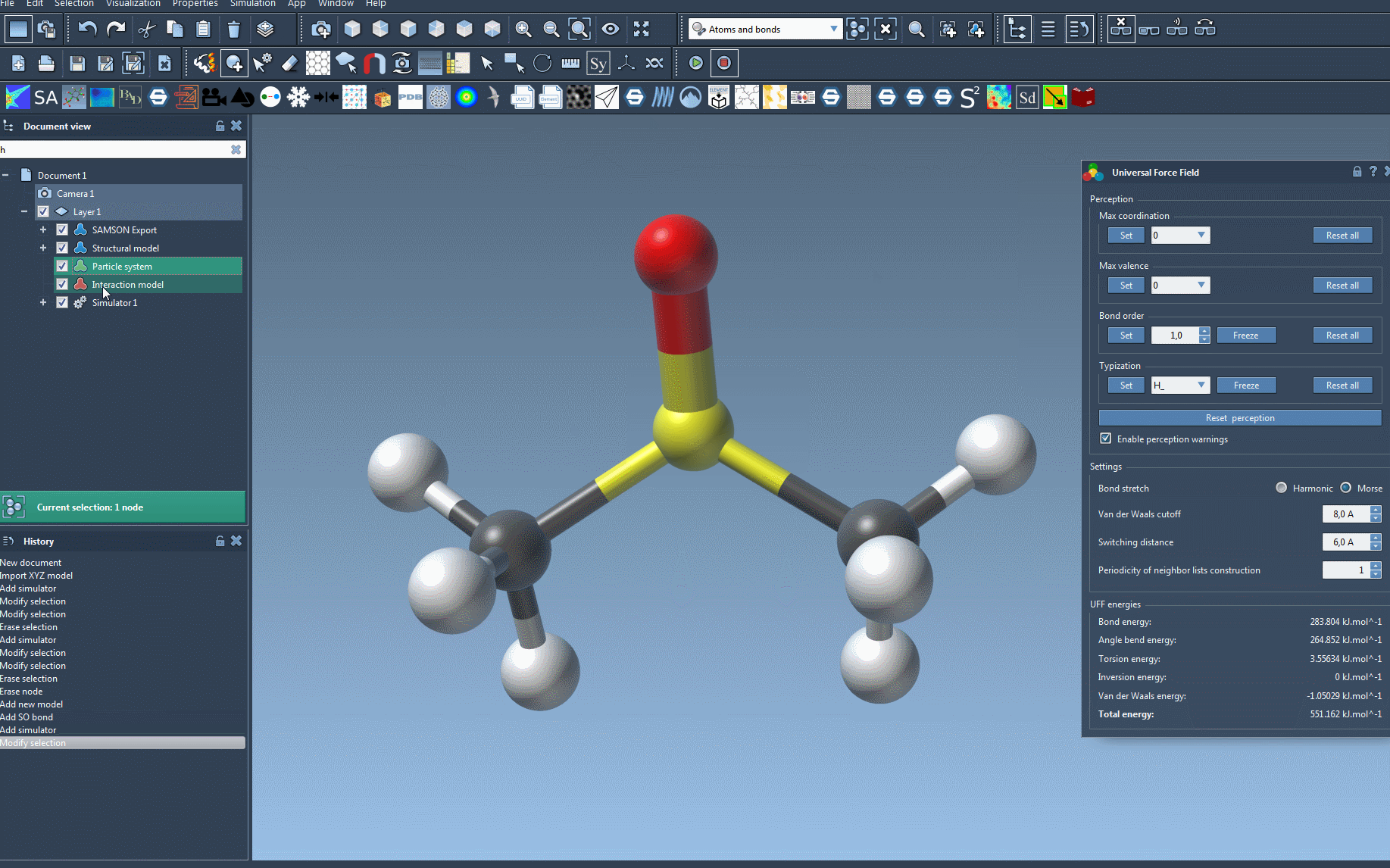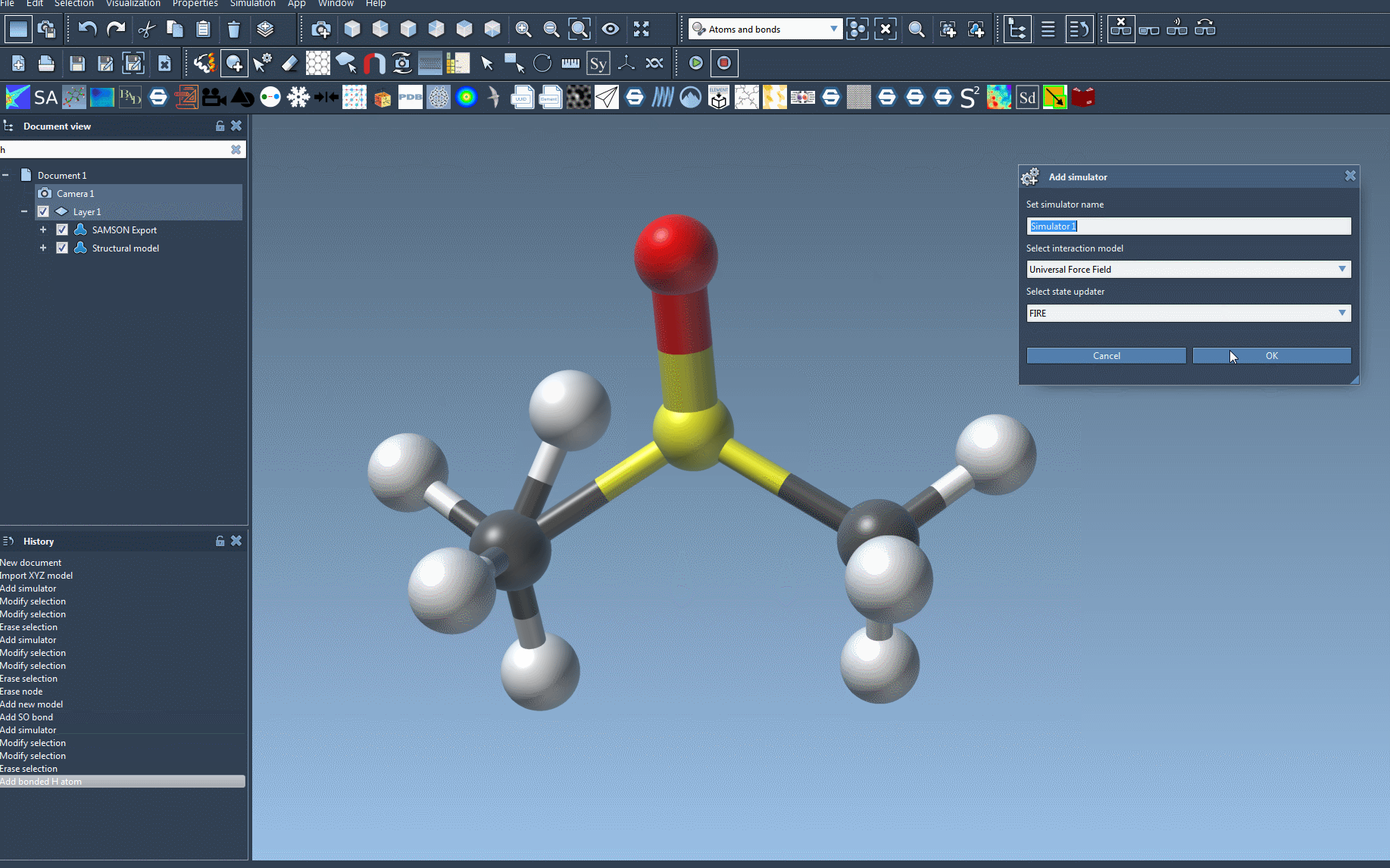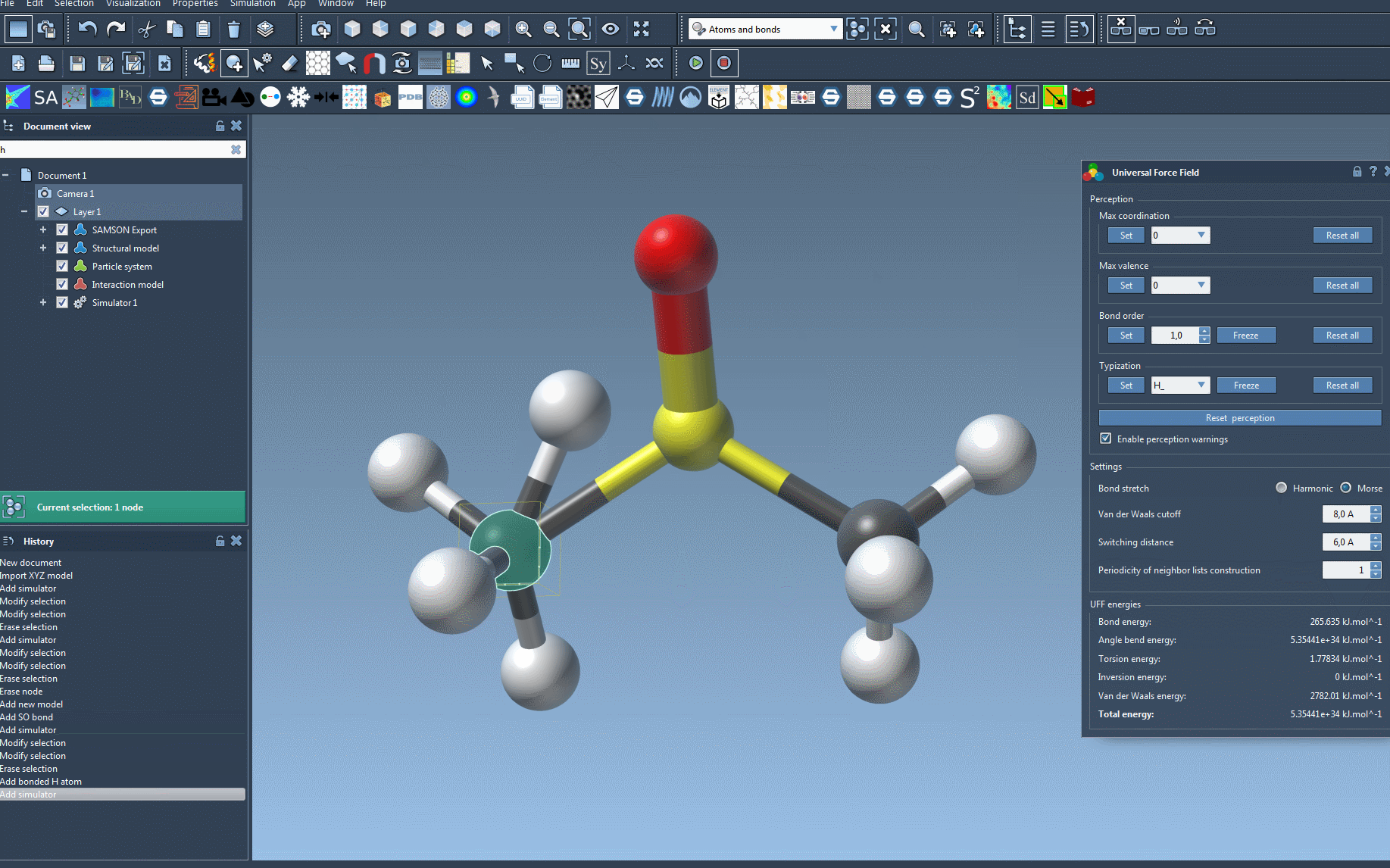
- Bond stretch interaction type: Choose between Harmonic and Morse.
- Van der Waals (vdW) cutoff: Set the distance beyond which vdW interactions are no longer considered.
- Switching distance: Define when vdW interactions start to smoothly taper off as they approach the cutoff limit.
- Periodicity of neighbor list construction: Optimize how often vdW neighbor lists are updated during the simulation.
Starting the UFF Simulation
After setup, launch the UFF simulation via Edit > Simulate > Start. The bottom portion of the UFF window displays a breakdown of energy contributions, including individual UFF terms and total energy. You can also interactively manipulate atoms and observe real-time adaptations in the molecular model.
Troubleshooting and Enhancing Your Workflow
For more advanced users, SAMSON allows refined control over UFF typizations and customization of parameters. This flexibility ensures that even rare or challenging configurations can be accurately modeled. Explore features like freezing bond orders or specifying atom types for additional precision.
By utilizing SAMSON’s automatic molecular perception capabilities and intuitive interface, you can transform a typically cumbersome process into a streamlined, efficient workflow. Whether you’re simulating molecular dynamics for research or industry, SAMSON’s UFF implementation empowers you to focus on results without getting bogged down by setup complexities.
For further details and advanced instructions, visit the official UFF documentation page at https://documentation.samson-connect.net/tutorials/uff/uff/.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.