





One of the challenges molecular modelers often face when preparing a system for simulations is defining the appropriate periodic box. If the box is too small, your system’s molecules might interact with their periodic images, leading to inaccurate results. On the other hand, unnecessarily large boxes can increase computational cost. This guide, based on SAMSON’s GROMACS Wizard documentation, explains how to efficiently prepare a periodic box that meets your simulation needs.
Why is the periodic box important?
Periodic boundary conditions (PBC) allow your simulation to mimic an infinite system by replicating the simulation box in all directions. To ensure accurate results, the box must be large enough to follow the minimum image convention, which states that molecules in the system should not interact with their neighboring periodic images.
Steps to define the periodic box in SAMSON
Using the GROMACS Wizard in SAMSON, you can define the box step by step:
1. Select the system
Select your molecular system from the Document view, or leave the selection empty if you want to include the whole system in your document.
2. Choose your box type
SAMSON supports several unit cell types:
- Cubic
- Orthorhombic
- Triclinic
- Rhombic dodecahedron
- Truncated octahedron
For most simulations, the Orthorhombic box is recommended due to its versatility. However, choose the box type that best suits your specific requirements. For more details, see GROMACS Wizard: Periodic boundary conditions.
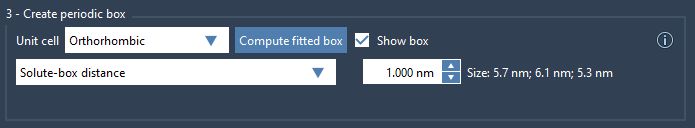
3. Define box dimensions
Two options are available for initial box fitting:
- Box lengths: Specify the box size directly. This ensures a uniform box size for batch projects with multiple conformations. You may need to adjust the size to ensure the minimum image convention.
- Solute-box distance: Specify the distance between the system and the box edges. At least 1 nm is recommended to satisfy the minimum image convention. This option automatically adjusts the box size for batch projects with varying conformations.
For this example, select the Orthorhombic box, then choose the Solute-box distance option and click Compute fitted box. This ensures the box dimensions fit your system with appropriate spacing.

4. Verify and adjust if necessary
Once the box is created, double-check to confirm that all atoms and molecules are comfortably within the box. Adjust the size if necessary, especially for large or highly flexible systems. Add at least 2 nm between periodic images of non-solvent molecules to avoid incorrect force calculations.
Tips and additional resources
If you’re simulating proteins or similar biological molecules, ensure the box dimensions account for possible conformational changes. This minimizes artifacts and ensures accuracy over time. For more information, check the minimum image convention guide in the documentation.
Final thoughts
Carefully setting up the periodic box is critical for accurate and meaningful molecular dynamics simulations, and SAMSON simplifies this process with its user-friendly GROMACS Wizard. With proper setup, you can avoid common pitfalls and ensure reliable results.
To learn more, visit the full documentation page: GROMACS Wizard – Step 1: Prepare.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON at https://www.samson-connect.net.