





One of the challenges in molecular modeling is managing smooth transitions during topology changes — like breaking or forming covalent bonds — without disrupting simulations. Introducing the Interactive Modeling Universal Force Field (IM-UFF), an extension of the Universal Force Field (UFF), designed to simplify such tasks with precision and efficiency. Let’s explore how IM-UFF enables intuitive handling of topology changes while simulating or editing molecular systems.
What Makes IM-UFF Unique?
The IM-UFF interaction model extends the capabilities of the UFF by enabling real-time changes to molecular topologies. This means you can create and break covalent bonds, modify bond orders, and adjust atom typizations dynamically. Whether you’re building, editing, or exploring molecular systems, IM-UFF allows significant modifications while staying guided by physically-based interatomic forces.
This feature is particularly useful in situations requiring smooth transitions between molecular states, such as during drug design or material science investigations. By allowing molecules to autonomously adapt their topology to reflect the local atomic organization, IM-UFF provides a pathway to more accurate and user-friendly molecular modeling.
Getting Started with IM-UFF
Here’s how to set up and work with IM-UFF:
- Ensure you have the IM-UFF Extension installed in your SAMSON workspace.
- Open a document with the molecular system you wish to simulate.
- Add a simulator by navigating to Edit > Simulate > Add simulator or using the shortcut
Ctrl+Shift+M(Cmd+Shift+Mon macOS). - Select Interactive Modeling Universal Force Field from the list of interaction models.
- Choose a state updater, like FIRE (Fast Inertial Relaxation Engine), and press the OK button.
Unlike conventional UFF implementations, there’s no setup window in IM-UFF; it’s ready to use immediately. Its parameter window offers a seamless integration of interactive modeling options, setting you up for efficient molecular simulations.
Transform Your Molecular Simulations
IM-UFF offers two noteworthy settings that provide flexibility in simulations:
- Static topology (UFF only): This option toggles between standard UFF (when checked) and IM-UFF (when unchecked).
- Keep vdW for manipulated: When active, van der Waals forces are fully accounted for. When turned off, manipulated atoms are excluded from vdW interaction calculations, making it easier to form new bonds without interference from repulsive forces.
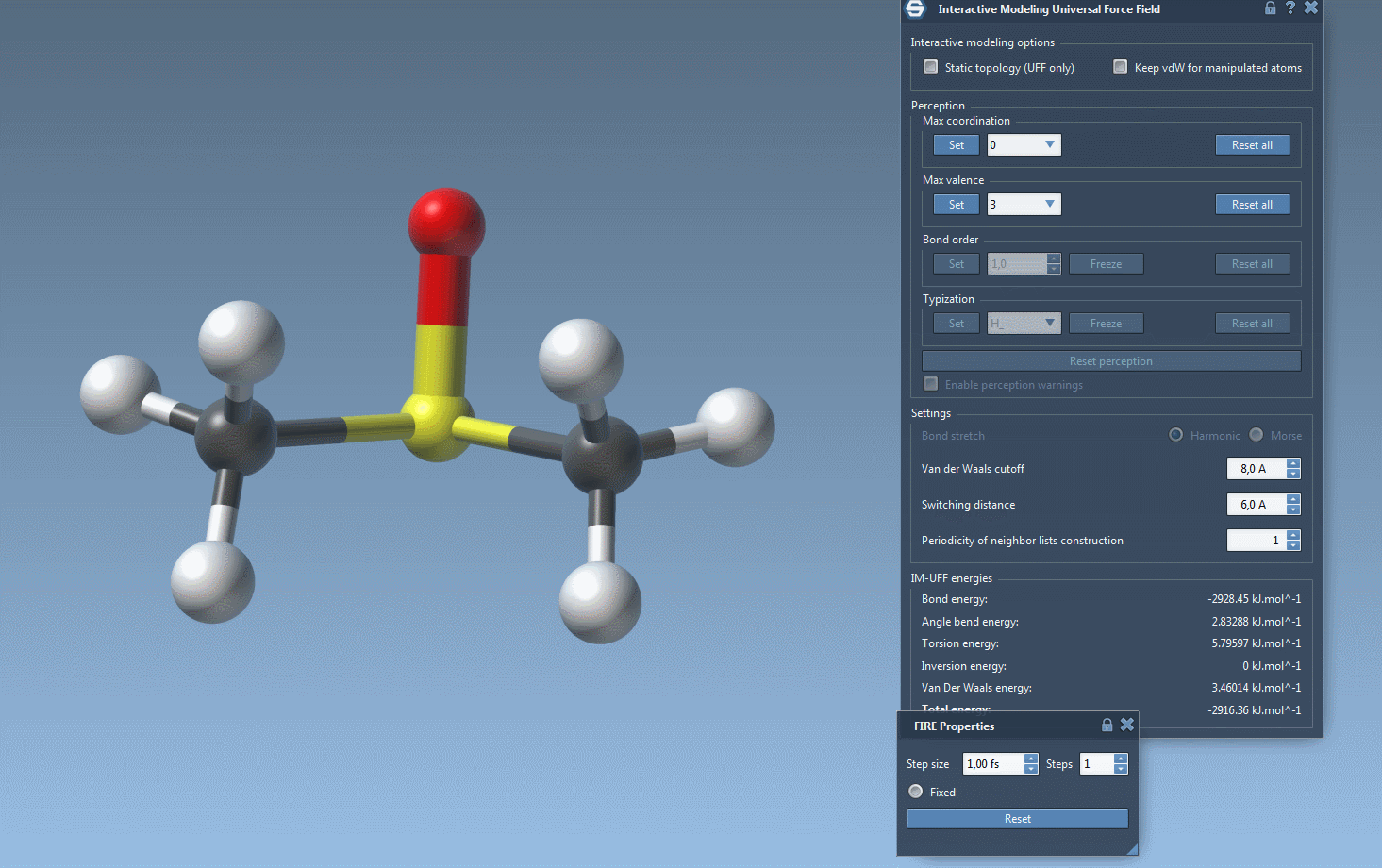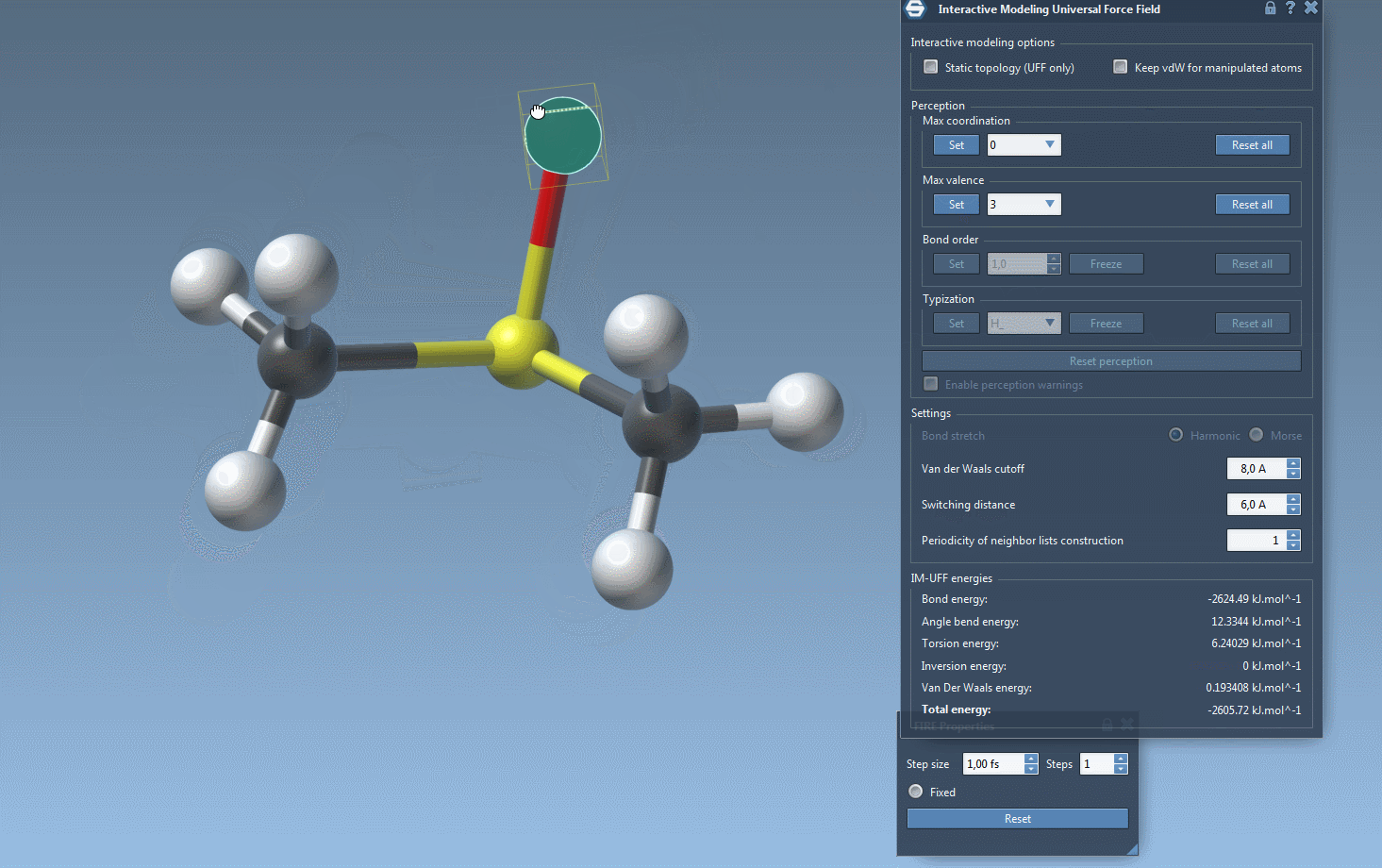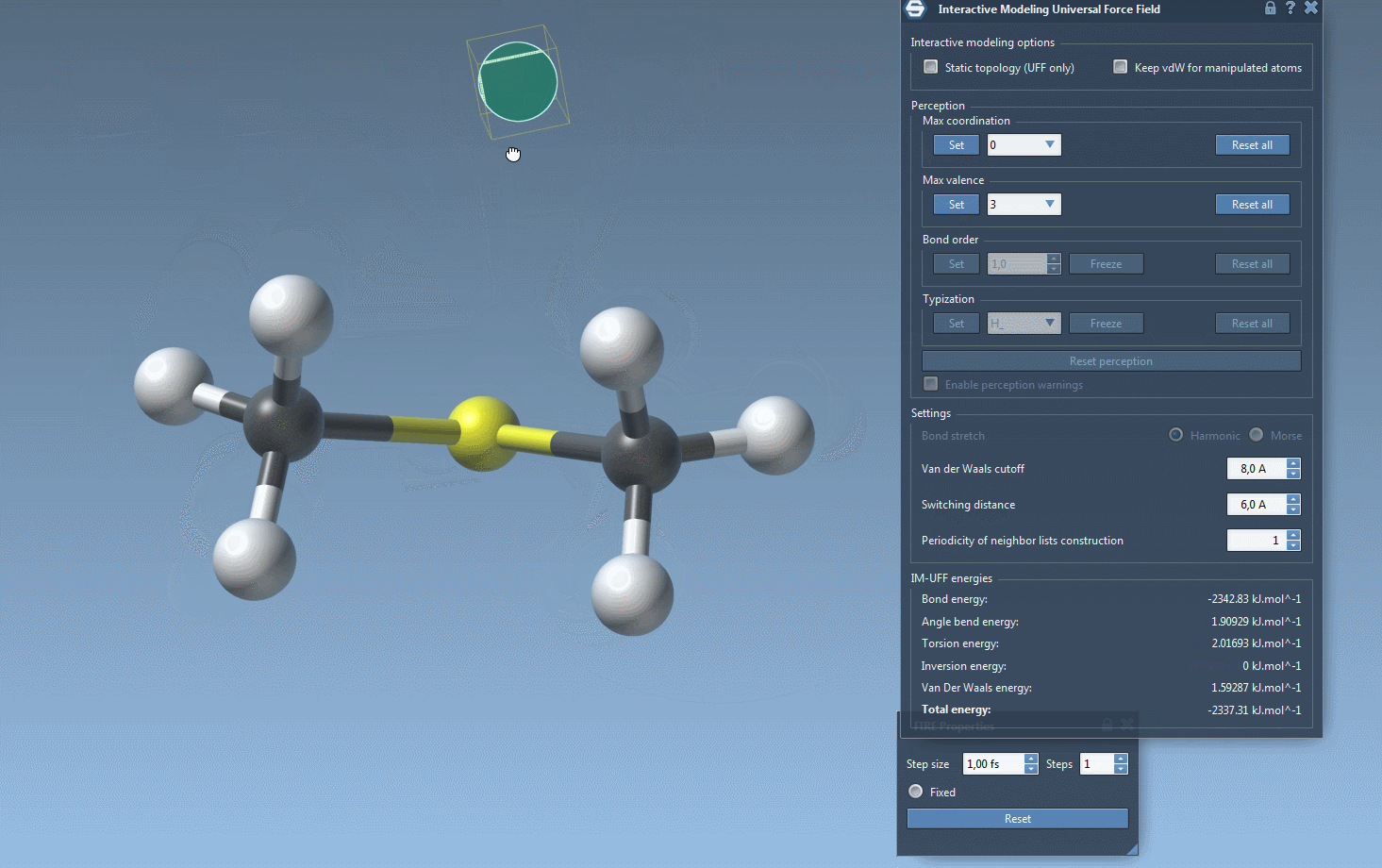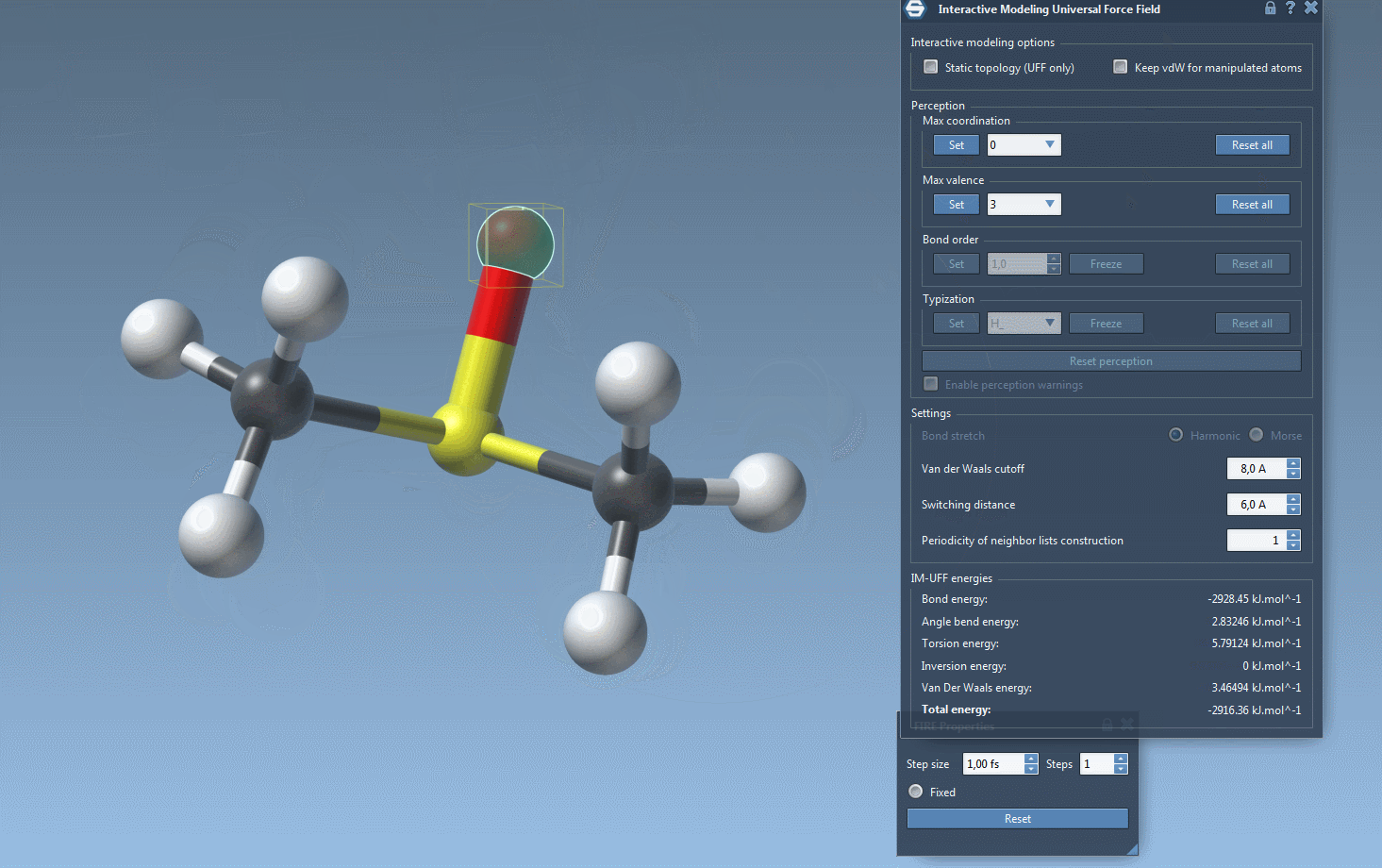
Run the simulation by ensuring both options are unchecked, then starting via the Edit > Simulate > Start button. Once active, you can observe the total energy of the system and its individual energy components in the parameter window.
As you interact with atoms by dragging them with your mouse, you’ll notice how bonds adjust dynamically. Small movements lead to structural adjustments, preserving topology, while larger displacements may lead to bond breaking and a subsequent topology update. Similarly, bringing atoms closer together can trigger the formation of new bonds, reflecting the new configuration.

Customization Options
IM-UFF also offers several customization options for advanced users. For example, you can adjust van der Waals cutoff settings, switching distances, and neighbor list periodicity. These settings allow more granular control over simulations, tailoring the interaction forces to the specific needs of your molecular system.
Furthermore, while running IM-UFF (static topology unchecked), bond orders and typizations are updated continuously based on atom positions, ensuring smooth topology transitions. You can also delete and add atoms dynamically, with the typization updating automatically for a flexible and efficient modeling experience.
Experience Smoother Molecular Modeling
IM-UFF revolutionizes how molecular modelers handle topology changes, making it easier than ever to simulate complex transformations intuitively. Whether you’re a researcher exploring novel materials or a professional working in drug discovery, IM-UFF offers a robust and interactive solution to improve your simulations.
For a complete guide, visit the full documentation page at this link.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Download SAMSON at https://www.samson-connect.net.