





For molecular modelers, understanding the Potential of Mean Force (PMF) is essential when studying biomolecular interactions. Yet, calculating PMF can often feel tedious, especially when dealing with complex simulation setups. Fortunately, the GROMACS Wizard in SAMSON offers a streamlined and efficient way to carry out PMF analysis using the Weighted Histogram Analysis Method (WHAM). This blog post walks you through a practical solution to this common modeling pain point.
Why PMF Analysis Matters
The Potential of Mean Force provides profound insights into biomolecular interactions by quantifying free energy changes along a reaction coordinate. It’s a fundamental method for studying binding affinities, conformational changes, and molecular pathways. However, performing WHAM-based PMF analysis often requires manually organizing input data, setting reaction coordinates, and interpreting results—steps prone to bottlenecks in the workflow.
Bringing Clarity and Efficiency to PMF Computation
The GROMACS Wizard makes the PMF computation process simpler and more accessible. Let’s take a closer look at how to get started:
1. Organize Your Input Data
The first step is properly structuring your project directory. Typically, you’ll be working with results from an Umbrella Sampling simulation. Organize your data into numbered subfolders, where each folder corresponds to results from simulations of the same system but different reaction coordinate values:
Even if this sounds like a tedious step, the GROMACS Wizard’s auto-fill button (![]() ) can save time by filling in the appropriate paths automatically if you’ve just completed the previous Umbrella Sampling simulation in the same project.
) can save time by filling in the appropriate paths automatically if you’ve just completed the previous Umbrella Sampling simulation in the same project.
2. Configure WHAM Analysis Parameters
After organizing your input directories, switch to the WHAM Analysis tab in GROMACS Wizard. Here, you can configure details such as:
- Reaction Coordinate: Select the reaction coordinate from a generated list.
- Bounds and Units: Optionally, you can set custom bounds and choose different energy units to suit your analysis needs.
- Computation Settings: Define parameters like custom timestamps for the analysis.
Once you’re ready, hit the Compute button. Depending on trajectory size, computations may take seconds to a few minutes.
3. Analyze and Visualize Your Results
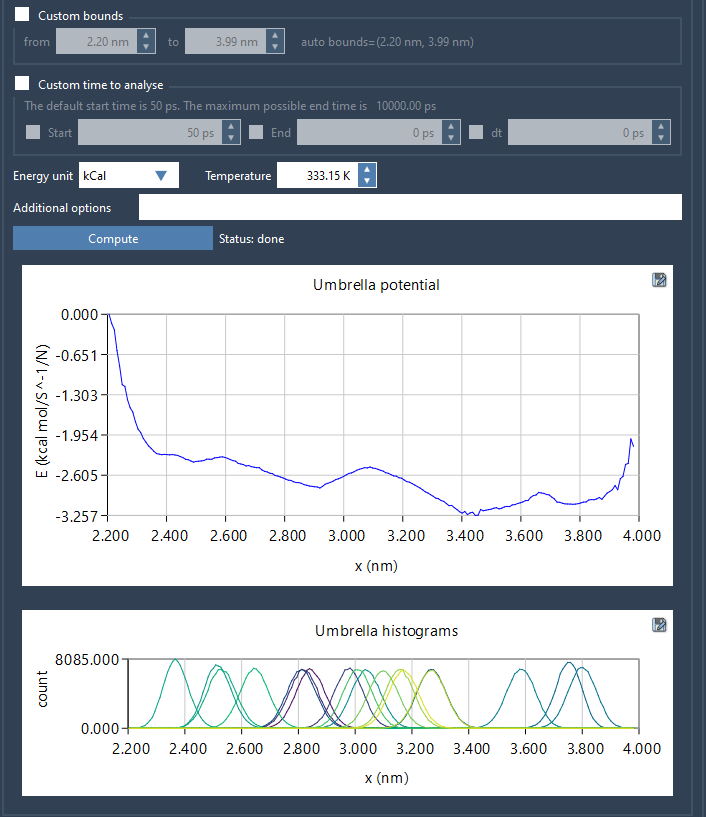
Once the computation completes, the GROMACS Wizard generates two graphical outputs:
- PMF Plot: This shows the free energy profile along the reaction coordinate.
- Histogram: This illustrates the coverage of your reaction coordinate space, helping you identify regions that might need additional simulations.
4. Save and Reuse Your Analysis
One of the most time-saving features of GROMACS Wizard is its ability to save results automatically in the wham_results subfolder of your project directory. If you explore multiple reaction coordinates, the tool reuses pre-computed data when you modify parameters. This auto-saving mechanism ensures efficiency, consistency, and easy reproducibility of your PMF computations.
Wrap-Up
The GROMACS Wizard in SAMSON offers a powerful yet user-friendly approach to streamlining the previously complex process of WHAM-based PMF analysis. By automating data input, empowering custom configurations, and visualizing outputs effectively, the tool minimizes time spent on tedious tasks and maximizes insights into molecular interactions.
To dive deeper into this workflow, consult the full documentation at this link.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here: https://www.samson-connect.net.