





Streamlining molecular simulations requires selecting the right tools and frameworks that adapt seamlessly to your systems. A frequent challenge for molecular modelers lies in properly applying trusted interaction models like the Universal Force Field (UFF) to their systems. This blog post will break down how to set up UFF step-by-step in the SAMSON platform for a smooth and efficient simulation process.
Introduction to Universal Force Field (UFF)
UFF is a computational tool designed to calculate molecular interactions for a wide diversity of systems. Proposed by Rappe et al., it covers the entire periodic table, making it versatile and widely used in molecular mechanics and dynamics. SAMSON takes this a step further by integrating UFF directly and automating tasks like perceiving molecular systems, computing bonds, bond orders, and atom types.
How to Set Up UFF in SAMSON
Follow these steps to use UFF for your molecular simulations:
- Open your molecular system: Start by loading a document with the molecular system you wish to simulate. SAMSON ensures precision when applying UFF to various structures.
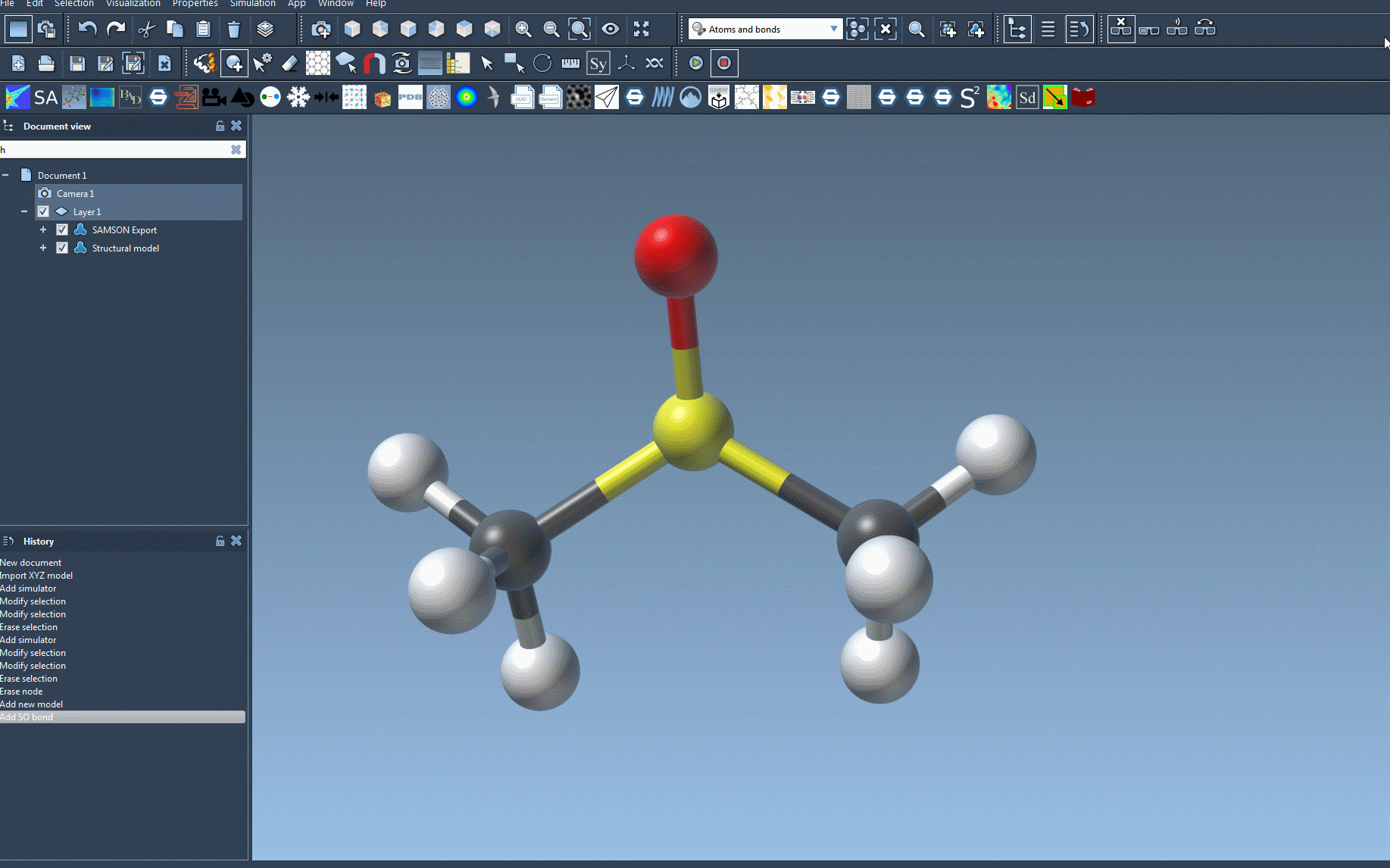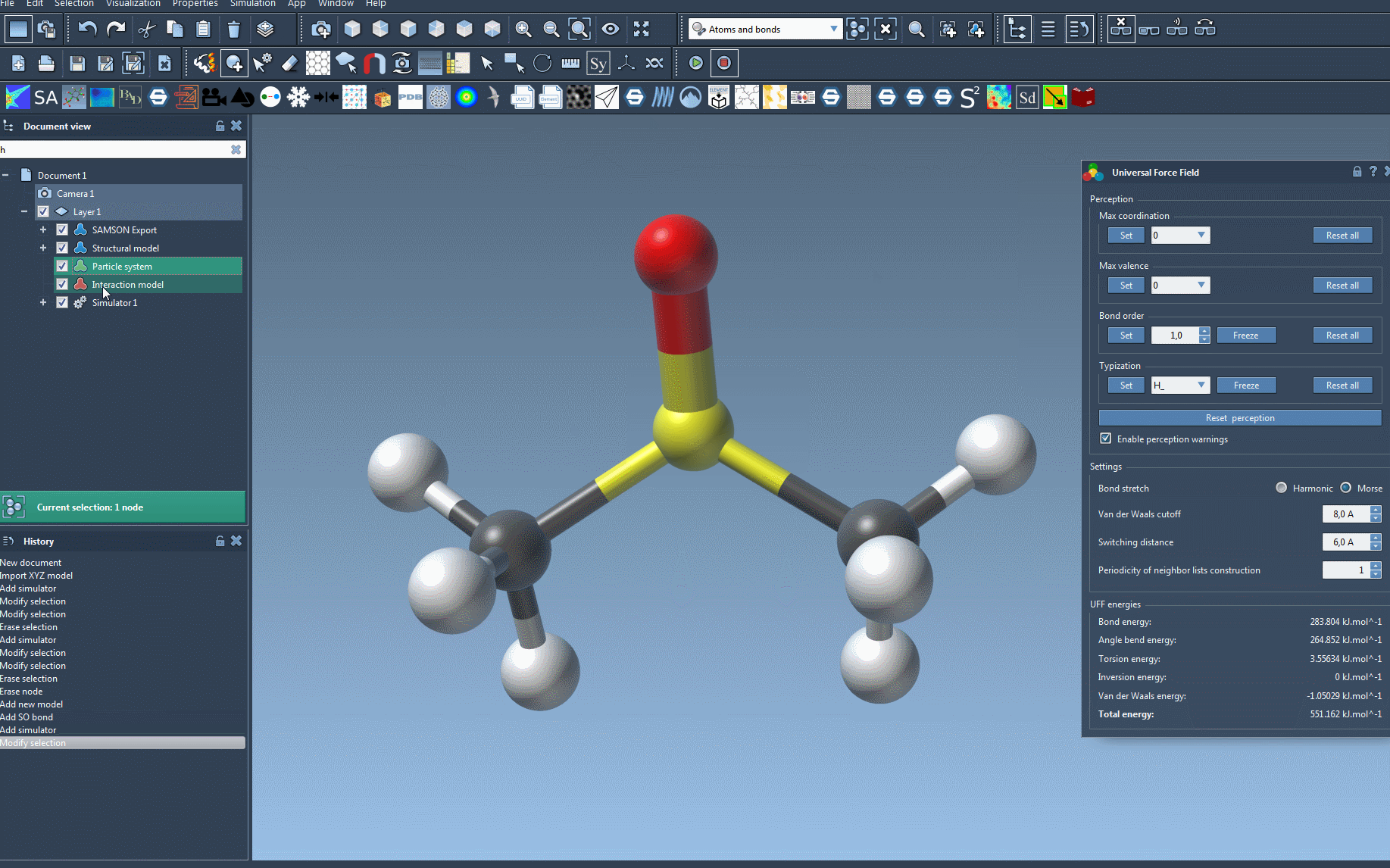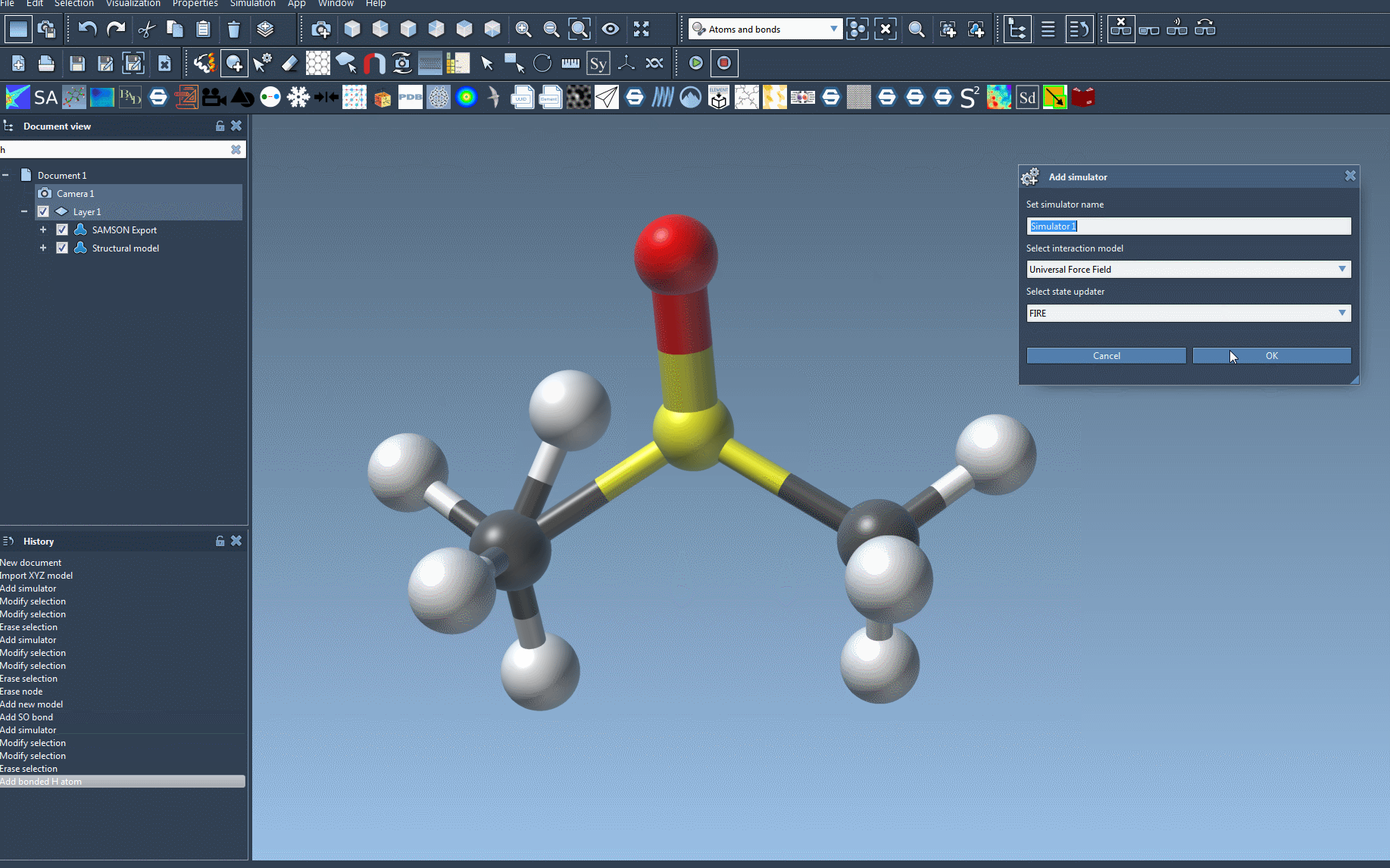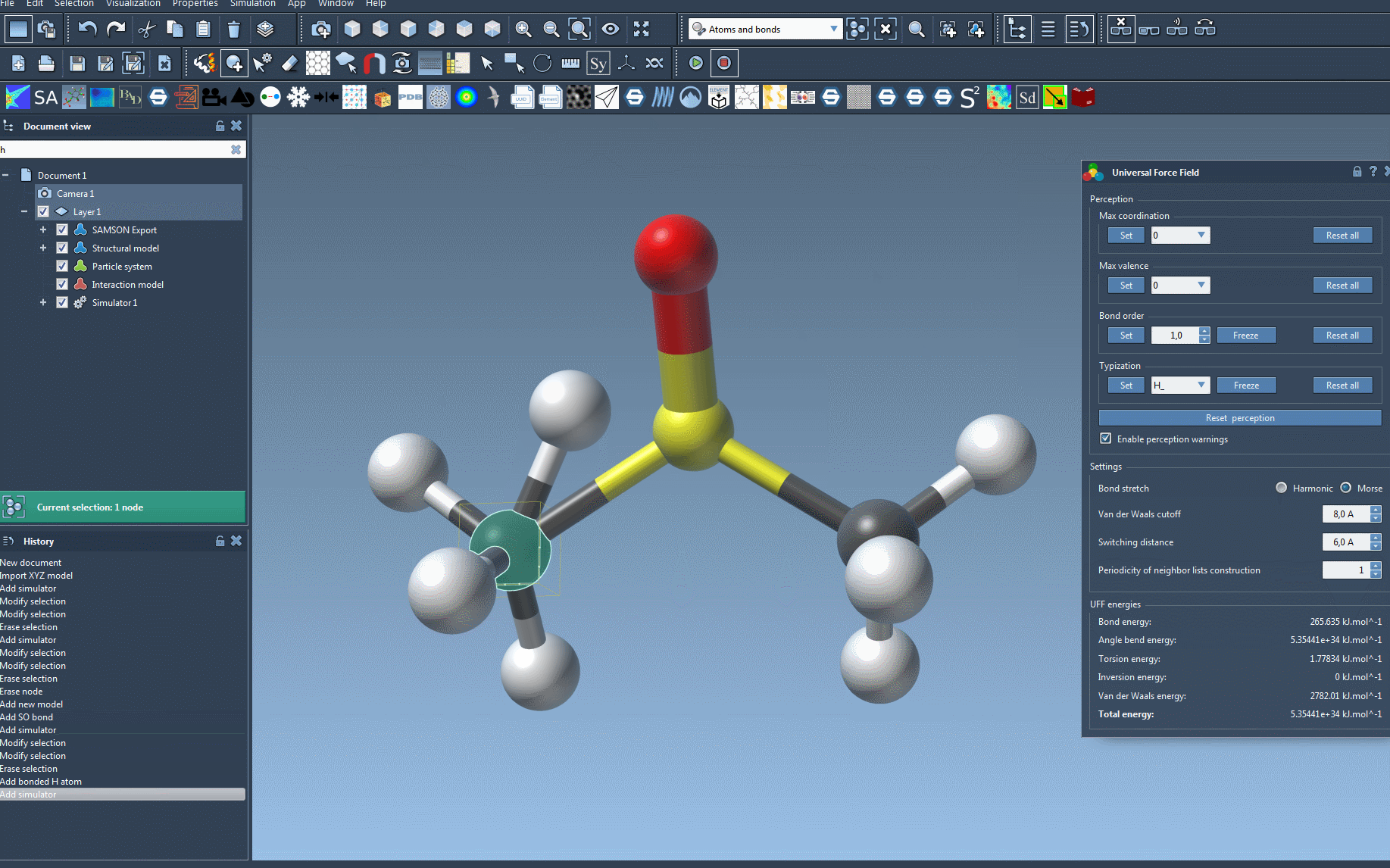
- Add a Simulator: Use the menu option
Edit > Simulate > Add simulatoror the convenient shortcutCtrl + Shift + M(orCmd + Shift + Mon Mac). - Select UFF: Choose ‘Universal Force Field’ from the list of interaction models available in SAMSON.
- Choose State Updater: Specify a compatible state updater for your simulation. SAMSON provides options like FIRE (Fast Inertial Relaxation Engine), allowing for smooth energy relaxation and optimization of your molecular system.
- Press OK: Confirm your choices, and the UFF setup window will appear to finalize the configuration.
Customizing the UFF Setup
The setup window gives you flexibility to refine your simulation preferences:
- Use Existing Bonds: Check this option if you’d like to rely on existing bonds in your model rather than recalculating them.
- Automatic Initialization: SAMSON computes essential parameters like bond orders, atom types, and covalent bonds (if existing bonds aren’t used).
Once initialized, you can modify various parameters, like the type of bond stretch interaction (Harmonic or Morse), van der Waals cutoff distances, and neighbor-list periodicity. These customizations allow tailored simulations based on the system’s structural needs.
Ensure a Trouble-Free Setup
If SAMSON detects any inconsistencies during initialization, it will provide warnings to guide you in resolving them. Afterward, you can proceed to the parameter window to further tweak and refine settings based on simulation objectives.
By following these guidelines, you will enable UFF to handle intricate molecular systems with confidence and precision.
To explore more details about UFF, including advanced typization and running simulations, visit the full tutorial at UFF Documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.