





For molecular modelers working with the Universal Force Field (UFF) to simulate molecular interactions, SAMSON offers straightforward and user-friendly tools to get started. This guide will walk you through setting up and configuring UFF simulations within SAMSON efficiently. From perceiving atom types and bonds to fine-tuning parameters, learn how to optimize your molecular simulations without unnecessary friction. Let’s address some of the common pain points that often occur during molecular system preparation.
Introduction to UFF and Molecular Simulations
Before diving in, it’s worth recalling that the Universal Force Field (UFF), proposed by Rappe et al., is a comprehensive force field based on molecular mechanics principles. It applies to the entire periodic table, making it immensely versatile for simulations. SAMSON streamlines this process, including automatic atom and bond perception, ensuring accurate calculations for bond orders, atom types, and molecular interactions.
Step-by-Step: Setting Up a UFF Simulation
Follow these steps to initiate your UFF simulation:
- Open your molecular system: Load the molecular system you wish to simulate into SAMSON.
- Add the UFF simulator: Go to
Edit > Simulate > Add simulatoror use the shortcut Ctrl+Shift+M (Cmd+Shift+M on macOS). From the selection menu, choose the Universal Force Field. - Choose a state updater: For example, you might select the Fast Inertial Relaxation Engine (FIRE) for dynamics updates.
- Configure bonds and initialization: In the UFF setup window, decide whether to rely on existing bonds or let UFF recompute bonds based on atom positions and types. This step also involves computing covalent bonds, bond orders, and atom types to ensure accurate simulations.
Run the Simulation and Monitor Parameters
Once the setup is complete, running the UFF simulation is just as simple:
- Click
Edit > Simulate > Startto begin the simulation.
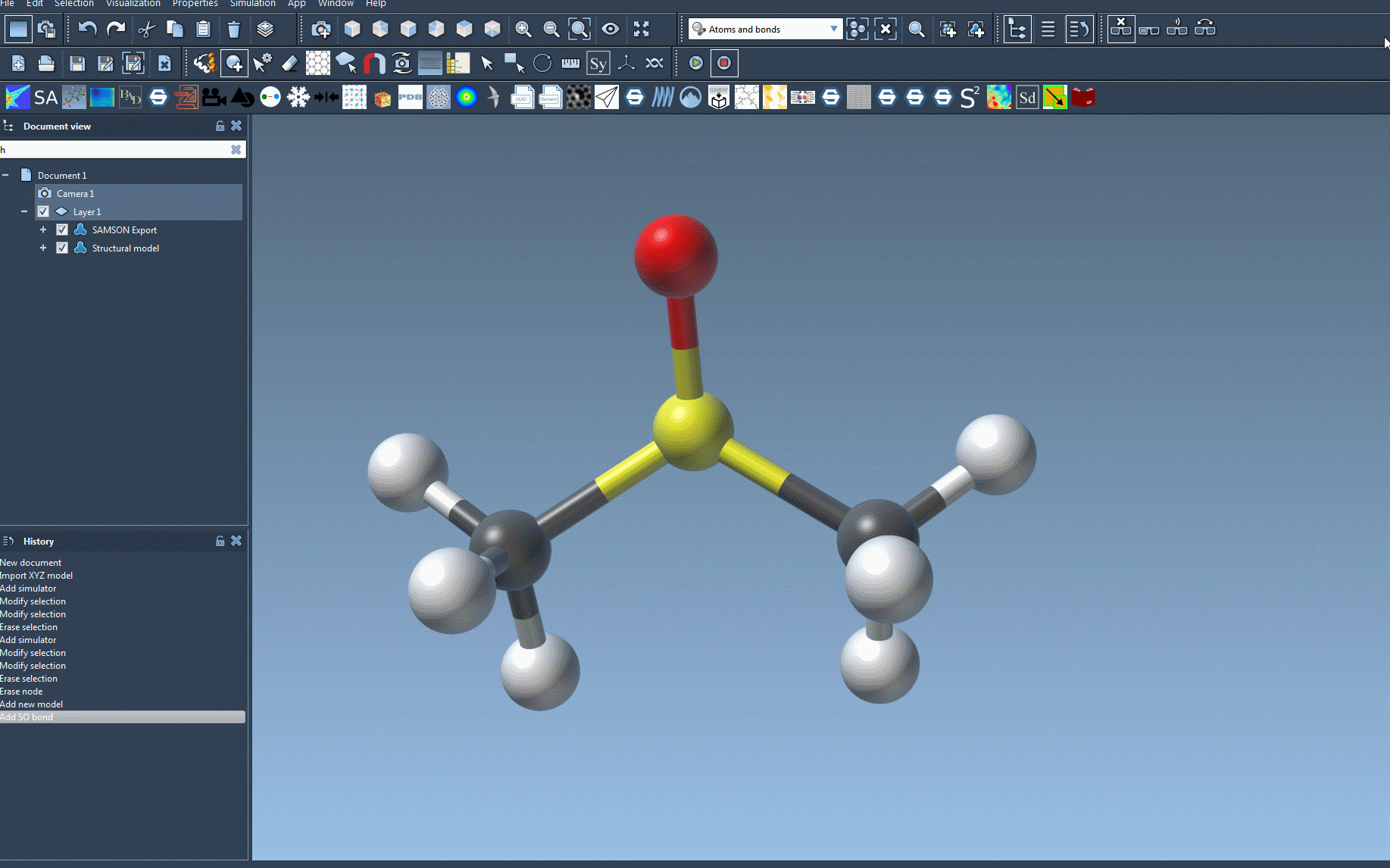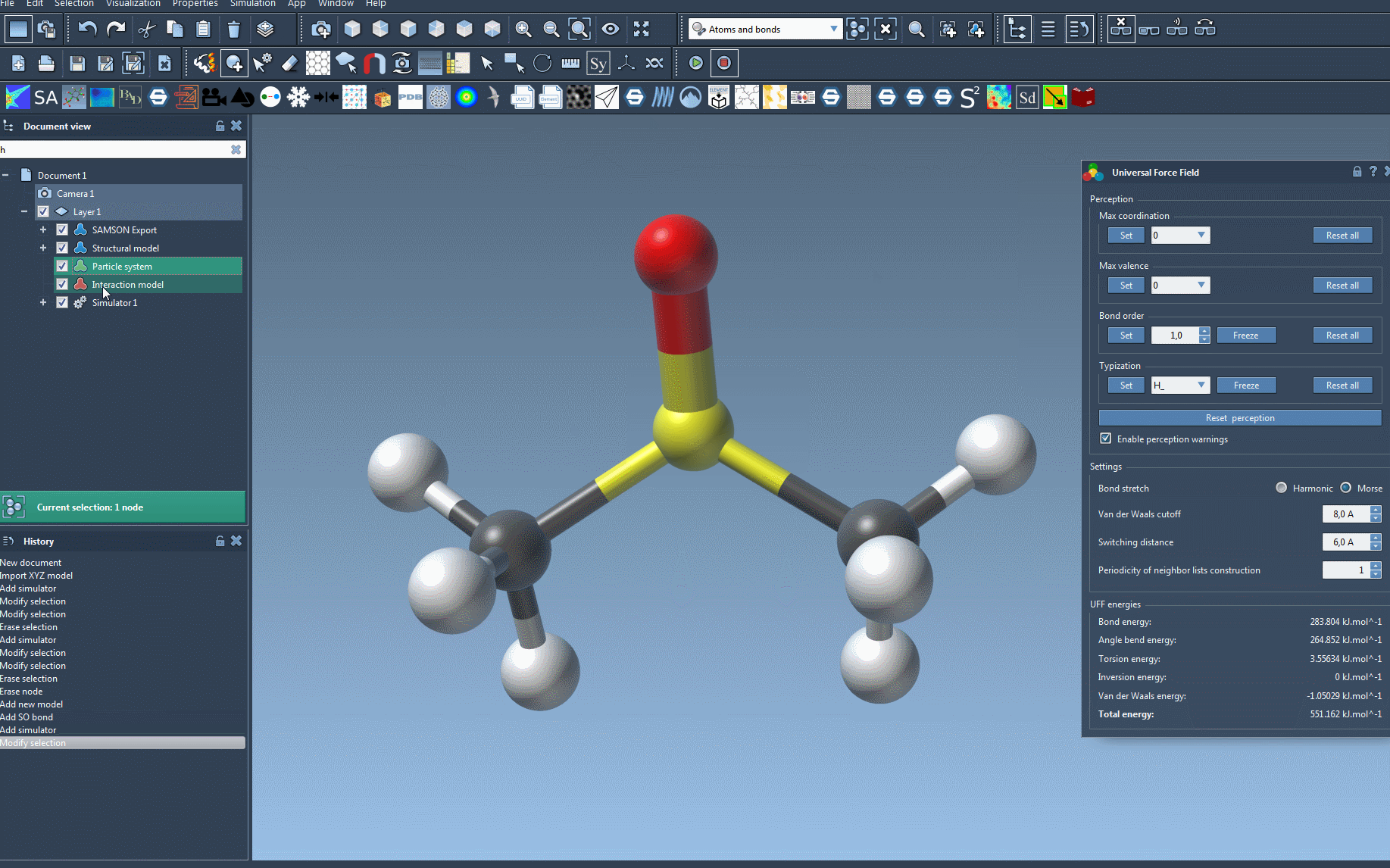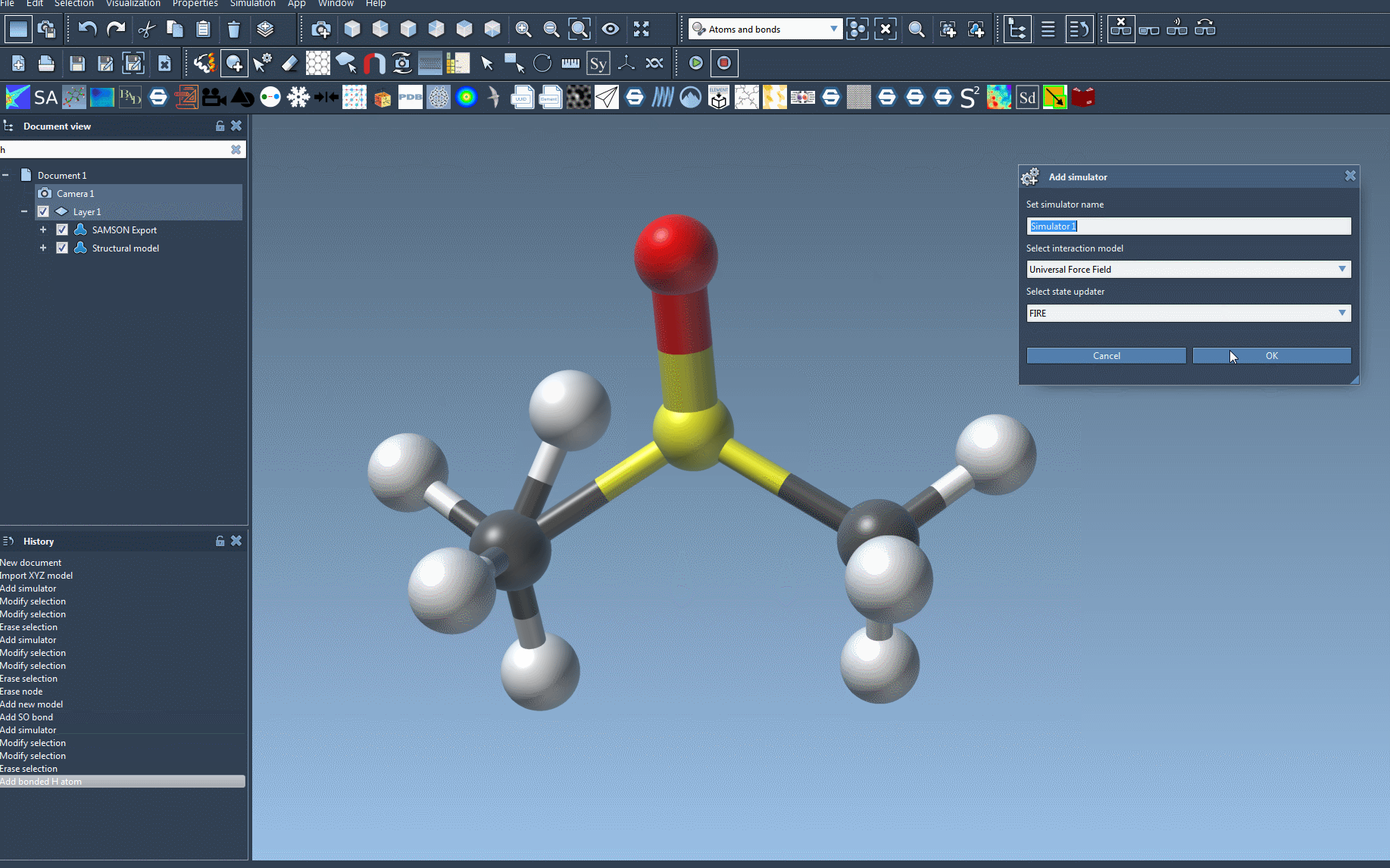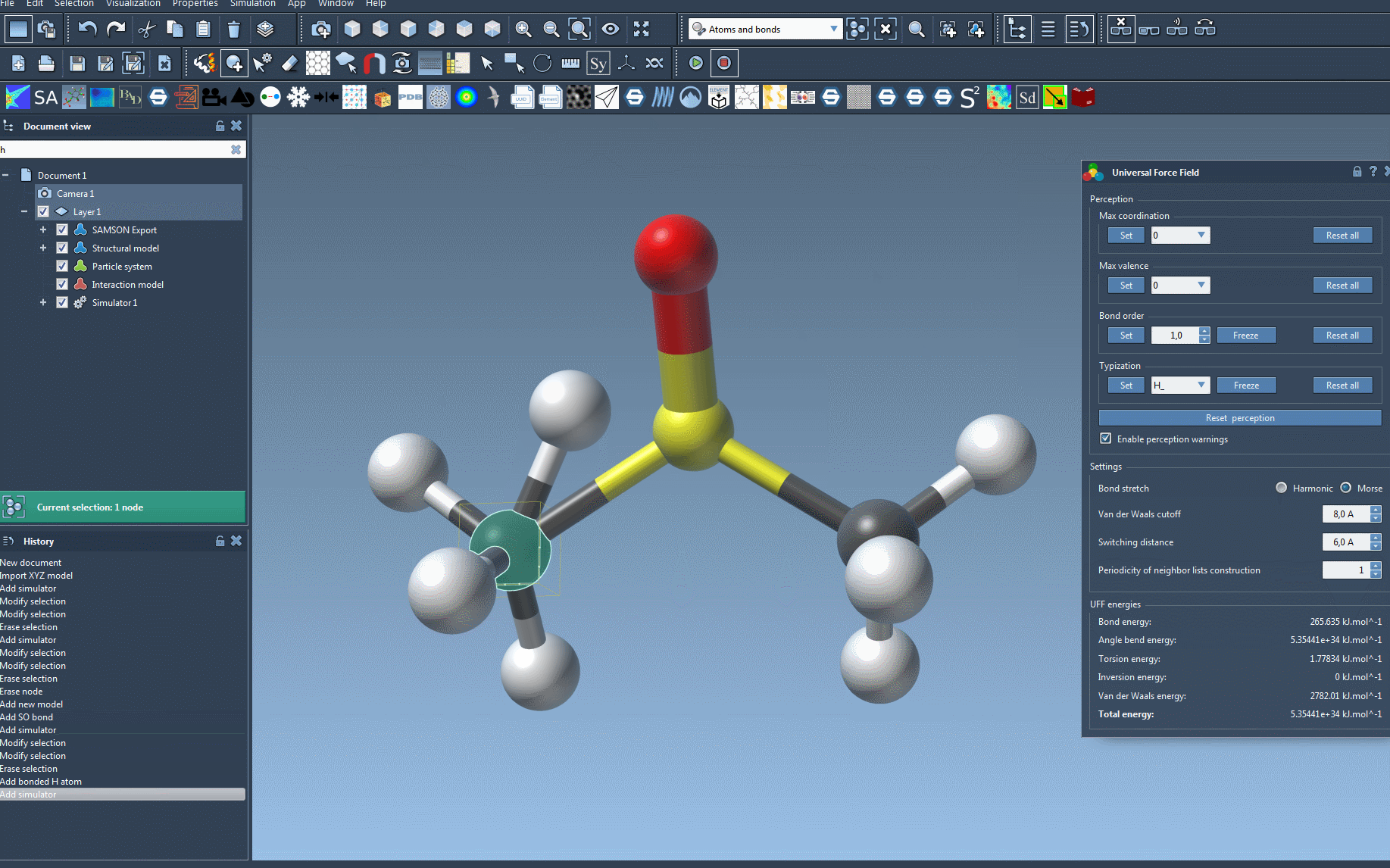
The UFF interface provides valuable insights into energy contributions, including the energies of specific UFF terms and the total energy. One exciting feature is that you can interactively move atoms within your molecular system and observe how the UFF model responds by updating the positions of neighboring atoms in real time.
You can also adjust key UFF parameters during the simulation, such as:
- Bond stretch interaction types: Toggle between Harmonic and Morse models for bond stretch calculations.
- Van der Waals (vdW) settings: Modify the cutoff distance and switching distance for vdW interactions.
- Neighbor list periodicity: Define how often neighbor lists are rebuilt during the simulation to balance performance and accuracy.
Why SAMSON Simplifies UFF Simulations
What sets SAMSON apart for UFF simulations is its ability to automate the complex steps of molecular system perception and configurations. For beginners or experts alike, this means less time spent on setup and more time spent analyzing meaningful simulation results. Whether you’re configuring bonds, customizing atom types, or adjusting cutoff distances, the platform ensures accessibility and precision with each interaction.
To learn more about setting up and running UFF simulations in SAMSON, refer to the original documentation available here.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.