





One of the frequent challenges faced by molecular modelers using umbrella sampling is the often cumbersome post-processing stage required to compute the Potential of Mean Force (PMF). The actual simulation might already have been computationally expensive, but without a streamlined and reliable way to process the output across multiple sampling windows, the final analysis can be time-consuming and error-prone.
This blog post walks through a particularly useful feature in GROMACS Wizard: the WHAM (Weighted Histogram Analysis Method) analysis for deriving PMF. Specifically, we’ll focus on how to organize your input data so that the Wizard can process it efficiently, with minimal user intervention.
Setting Things Up in GROMACS Wizard
To begin PMF analysis, switch to the WHAM Analysis tab within the GROMACS Wizard interface. A cleanly organized project folder is key. If you’ve previously performed an umbrella sampling simulation (such as via this tutorial), GROMACS Wizard can automatically infer your project path. Simply click on the auto-fill button:
![]()
Folder Structure: A Common Pain Point Made Simple
Your folder structure must conform to what GROMACS Wizard expects for batch processing: one main folder containing numbered subfolders, each representing a window of your umbrella sampling and containing the results for a simulation with a fixed reaction coordinate.
This simple structure is essential:
umbrella_sampling_project/000/001/002/- …
Here’s an example from the documentation that shows how your folders should be structured:
Once this structure is in place, GROMACS Wizard will scan the folders to extract information on reaction coordinate values, timing, and simulation temperature. The entire set of WHAM analysis options then becomes available. Users can select a specific reaction coordinate, define custom bounds or energy units if needed, and begin computation with a click.
What You’ll Get
Once computed, two plots are produced:
- A Potential of Mean Force (PMF) plot
- A histogram clearly showing the coverage of the reaction coordinate space
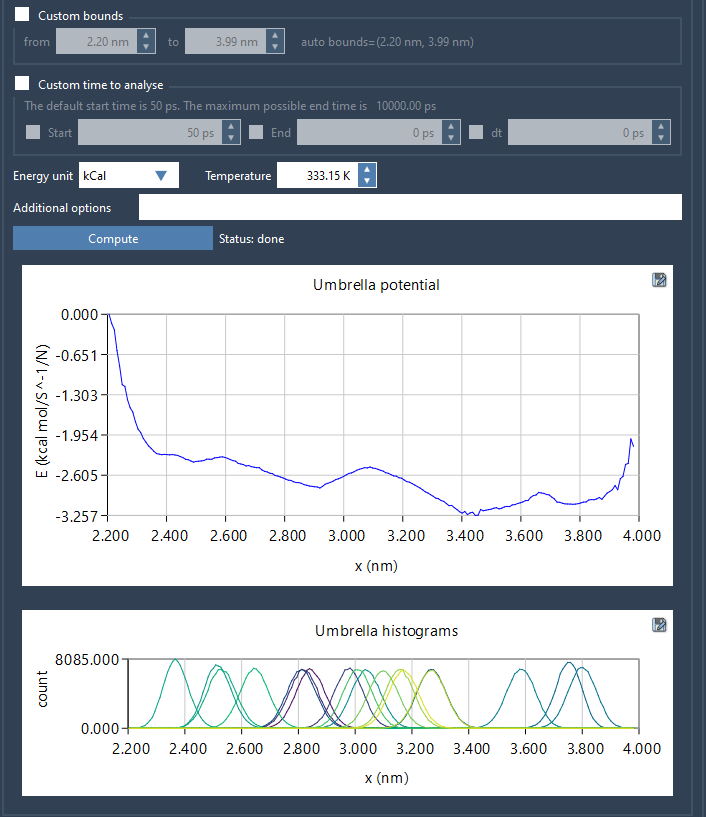
The histogram is particularly useful: it tells you where your sampling is sufficient—and where more runs might be necessary.
Saving and Reusing Results
Once computed, all results are stored automatically in a wham_results subfolder. Results are indexed by reaction coordinate and parameter settings, so if you tweak your inputs or switch coordinates, GROMACS Wizard can reuse existing computations—saving valuable time on long trajectories.
Whether you’re checking convergence or generating publication-quality graphs, having clearly organized output speeds every step of the process.
To learn more, visit the original documentation page: https://documentation.samson-connect.net/tutorials/gromacs-wizard/pmf-analysis/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON here.