





When modeling molecular systems, capturing the transition between two stable states can be crucial—whether you’re studying ligand unbinding, protein conformational changes, or reactions at the atomic level. Molecular modelers often face the challenge of refining the rough paths generated from interpolations or simulations so they’re physically meaningful and energetically accurate.
This is where the Parallel Nudged Elastic Band (P-NEB) method in SAMSON can help. P-NEB improves rough or approximate transition paths by using a physically motivated optimization that distributes intermediate states (or conformations) along a minimum energy path. It essentially “nudges” keyframes on the path using energy gradients, keeping them spaced like beads on an elastic band. 🧬
Why This Matters
Say you already created a transition using linear interpolation or SAMSON’s Ligand Path Finder. While these paths look smooth, they might not reflect the actual energetics of your system. P-NEB refines these paths using force-field-based optimization, giving you enhanced realism and greater confidence in the geometry of high-energy intermediates.
What’s more, SAMSON’s implementation allows parallel computation, meaning that it can refine each conformation independently, speeding up the workflow significantly if you’re working on a multi-core machine. ⚡
How to Do It
Once your system is set up in SAMSON and you’ve generated a path, here’s how to apply P-NEB refinement:
- Open the sample model by going to Home > Download and inserting one of the two provided molecule paths. For example: Zinc ligand unbinding trajectory.
- Go to Home > Apps > P-NEB or search for “P-NEB” in the Find everything… search bar.
- In the Document view, select a path node.
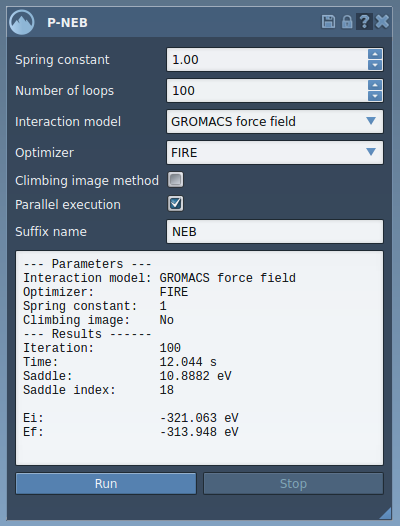
- Configure the P-NEB app settings as follows:
- Spring constant: 1.00
- Number of loops: 100
- Interaction model: Universal Force Field
- Optimizer: FIRE
- Climbing image: unchecked (optional)
- Parallel execution: checked
- Suffix: “NEB”
- Click Run to begin the optimization process. You’ll be asked whether to use existing bonds. Choose “Yes” and press OK.
Once the calculation finishes, a new, optimized path will be displayed in the Document view. You can double-click it to visualize or animate the refined trajectory. SAMSON also provides a convenient computation summary right within the app interface.
Prefer Paths Over Conformations
You can also apply P-NEB to a set of conformations. However, it’s recommended to combine conformations into a path first for faster performance. This can be done in the Document view by selecting your conformations and using the context menu: Conformation > Create path from conformations.
Final Thoughts
By refining interpolated or precomputed transitions with P-NEB, you make your simulations more physically realistic while saving time through parallel processing. Whether you’re studying a small ligand or a large molecular machine, this method makes it easier to elucidate energy landscapes with more confidence.
To dive deeper into the settings and examples, visit the full tutorial:
https://documentation.samson-connect.net/tutorials/pneb/optimize-transition-paths-with-parallel-nudged-elastic-band/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.