





Predicting the structure of protein-ligand complexes from NMR data often requires significant manual effort, especially when dealing with unassigned methyl peaks. The combinatorial nature of the problem can quickly become overwhelming: Which methyl belongs where? How do distance restraints from NOESY spectra constrain the system? If you’re a molecular modeler facing these challenges, there’s now an integrated way to simplify the process.
Enter NMR2 in SAMSON, an intuitive extension that helps you process NMR-derived restraints and predict possible binding poses—even when facing ambiguity in methyl assignments. Here, we highlight a specific use case that solves a familiar pain: defining and managing distance restraints for automated structure prediction via NMR data.
Why distance restraints matter
Distance restraints are at the heart of NMR-based structure determination. In NMR2, these restraints define allowable distances between atom pairs (or groups like methyls and pseudo-atoms), typically extracted from NOESY spectra. However, entering these restraints outside of a controlled interface often leads to format errors, mismatches, or inconsistencies. NMR2 offers a clean and streamlined interface to set them up with minimal risk of error.
How to specify distance restraints in NMR2
Once the receptor, ligand, and binding site are specified in SAMSON’s Document view, all NMR2 requires is a list of restraint lines in the form:
|
1 |
SITE_1 SITE_2 = LOWER_BOUND UPPER_BOUND |
Each line corresponds to a distance constraint between two sites such as hydrogen atoms, pseudo-atoms, or methyl groups. For instance:
|
1 2 3 |
Q4 M5 = 2.18 3.88 H8 M5 = 3.80 5.70 Q4 M1 = 3.73 6.64 |
Here, M5 represents an unassigned methyl group whose placement will be resolved during the automated prediction. The interface handles this just like other entries, requiring no additional format setup.
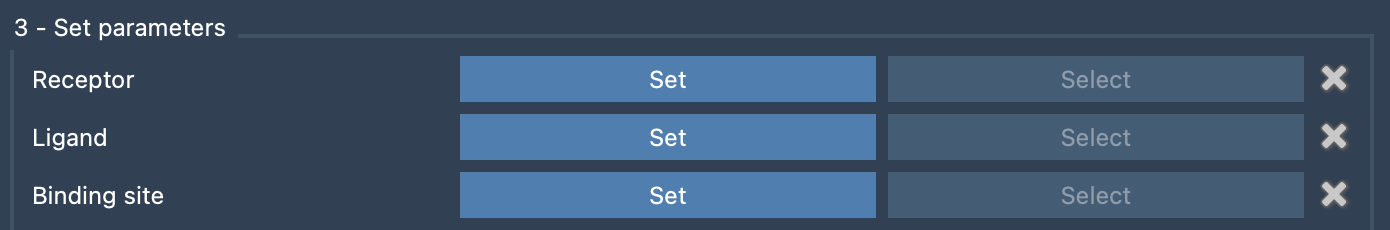
Adding known assignments to accelerate prediction
Have some methyls already assigned based on other experiments? Include them in the Assigned methyls box to reduce computation time and refine predictions. This is useful especially when the number of methyl groups involved starts increasing:
|
1 |
M5 = 130 QE |
This tells NMR2 that methyl #5 corresponds to the pseudo-atom QE on residue 130, helping the algorithm narrow the search space.
The benefit: Better predictions, less effort
Once everything is entered, click Predict structures. NMR2 will compute various putative complexes by testing multiple methyl arrangements against the provided restraints. The best-ranked predictions (based on how well they satisfy constraints and avoid steric clashes) can be imported into SAMSON for inspection and further analysis.
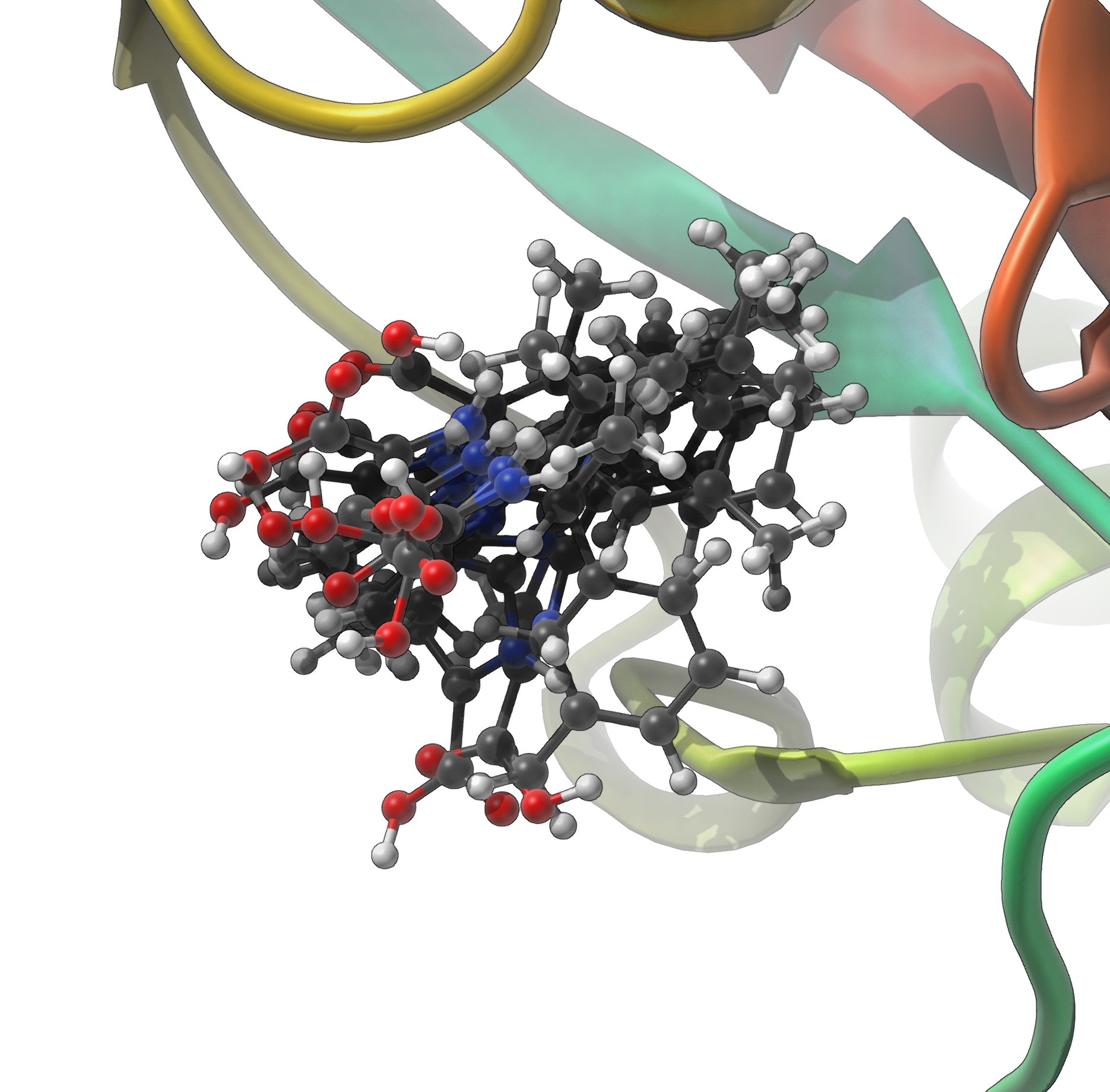
This approach eliminates the need for laborious scripting or file formatting. Instead, you’re free to focus on refining structural hypotheses and exploring functional implications.
Bonus: Aligning and inspecting predictions
All predicted complexes can be loaded together, cleaned of pseudo-atoms via a simple Document view filter, and aligned using alpha-carbons. This makes inspecting conformational variability (especially in ligand positions) fast and informative:
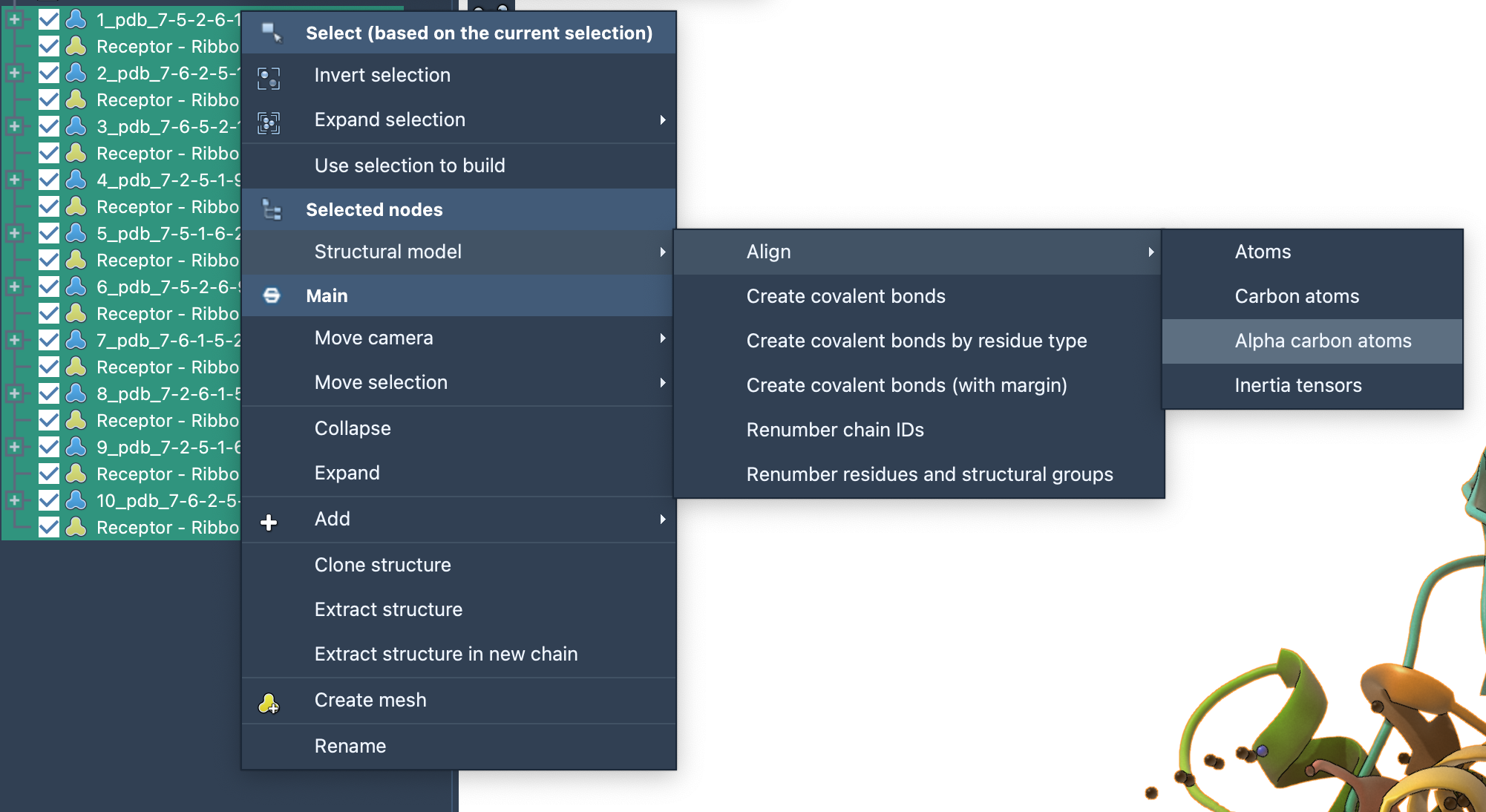
In a few clicks, you go from ambiguous NOESY peaks to visual structures of potential binding modes.
Curious to implement this structure prediction workflow in your own project? Explore the full tutorial here.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.