





Molecular modelers often need to refine rough transition paths when studying complex molecular systems. These paths, which connect important conformational states of molecules (e.g., ligands moving through proteins), can sometimes be physically inaccurate or unrealistic when first generated. This is where the Parallel Nudged Elastic Band (P-NEB) method comes in, offering an effective way to optimize such paths while maintaining even distribution of intermediary states.
If you’re working with SAMSON, the integrative molecular design platform, you’re in luck: the P-NEB app is specifically designed to help you perform these optimizations efficiently. This step-by-step guide will outline how to refine a transition path to create more accurate and meaningful molecular models.
What Does the P-NEB App Do?
The P-NEB app optimizes paths or sets of conformations to refine molecular transition paths. In simple terms, it iteratively adjusts intermediate states in the calculated pathway to ensure they respect the underlying physics, like energy landscapes, while keeping neighboring states evenly distributed using spring forces. This is crucial for applications like transition state analysis or ligand unbinding studies.
Key Settings in the P-NEB App
The P-NEB app offers several customizable parameters to tailor the optimization process:
- Spring constant: Controls the force keeping frames evenly distributed. Try starting with 1.00.
- Number of loops: Adjust this to define the number of optimization steps, for example, 100 iterations.
- Interaction model: Choose the force field used to compute energies and forces. “Universal Force Field” (UFF) is a general-purpose option.
- Optimizer: The optimization algorithm, with “FIRE” (Fast Inertial Relaxation Engine) being the default choice.
- Climbing image method: When active, focuses attention on the highest-energy points for finding saddle points. Useful for detailed studies but not needed in simpler cases.
- Parallel execution: Enables multi-threading for faster optimization.
- Suffix name: Adds a name to the output, e.g., “NEB” for clear labeling.
How to Optimize a Path
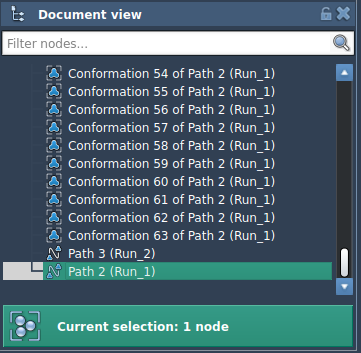
1. In your SAMSON document, select a path node from the Document view.
2. Open the P-NEB app (Home > Apps > All > P-NEB).
3. Customize the settings according to your requirements (see the setting details above), and then click Run.
4. During initialization, you’ll be prompted to confirm whether to use existing bonds in the “Universal Force Field”. Confirm this and proceed.

5. The status bar will show the optimization progress, offering live feedback on computation:

6. Upon completion, you’ll see the optimized path in the Document view:
Analyzing the Results
After optimization, the app provides a summary of the computation:
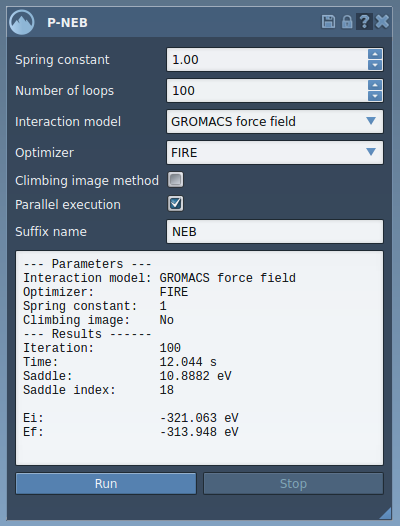
You can examine the new path by selecting it in your document. Use the Inspector to explore more details, or double-click to animate the transition. Right-clicking offers additional options to modify the path or export details for further analysis.
Why Use P-NEB?
The P-NEB method is particularly valuable for:
- Refining ligand unbinding pathways, where initial paths often lack accuracy.
- Transition state studies, where even small inaccuracies in energy trajectories can mislead conclusions.
While it’s easy to treat the initial pathway as “good enough,” employing P-NEB ensures your molecular simulations rest on solid ground.
For additional details and guidance, visit the official documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.