





Researchers often need to visualize or quantify conformational transitions in proteins. Whether you’re modeling domain movements, setting up umbrella sampling, or preparing steered dynamics, having a continuous path between two experimentally determined structures can save you time—and avoid launching time-consuming simulations from scratch. This post illustrates how to generate such a path using the ARAP Interpolator in SAMSON.
The As-Rigid-As-Possible (ARAP) interpolation algorithm constructs smooth, geometry-preserving transition paths. Instead of simulating dynamics, it computes an interpolated trajectory that maintains local structure as much as possible, making it a valuable tool for modelers preparing workflows or visualizations involving protein motion.
Why it solves a pain
Let’s say you have two crystal structures of the same protein in different functional states. Maybe one is ligand-bound, another not. You want more than two snapshots—you need a transition path. Simulations such as MD or enhanced sampling methods are reliable but often cost you extensive setup time, computational resources, and parameter tuning. ARAP interpolation builds the transition geometry in seconds, giving you a series of plausible intermediates that preserve biological realism and can serve as inputs to other modeling or simulation steps.
The basics: pick your start and goal
Here’s how to create a simple transition path between two conformations of the same protein using ARAP.
In this tutorial, we use two conformations of the Diphtheria Toxin—1DDT and 1MDT—from the Protein Data Bank:
- 1DDT, chain A: starting structure
- 1MDT, chain A: target conformation
First, load them via Home > Fetch in SAMSON by typing:
|
1 |
1DDT 1MDT |
Make sure to delete chain B from 1MDT to focus the interpolation on chain A only. Clean the structures using Home > Prepare to remove water, ligands, and alternate locations. It’s essential since ARAP requires both structures to form connected components.
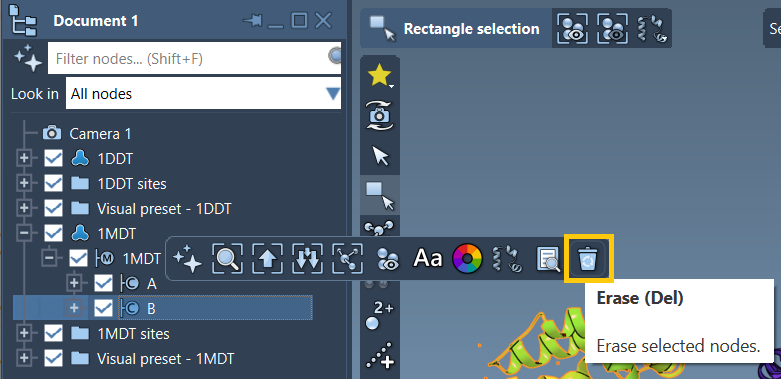
Create conformations for interpolation
In SAMSON, a conformation is a labeled structure used as input/output of applications that require multiple protein states.
- Select
1DDT, then go to Edit > Conformation and name it1DDT A. - Repeat for
1MDTand name it1MDT A.
Run ARAP Interpolation
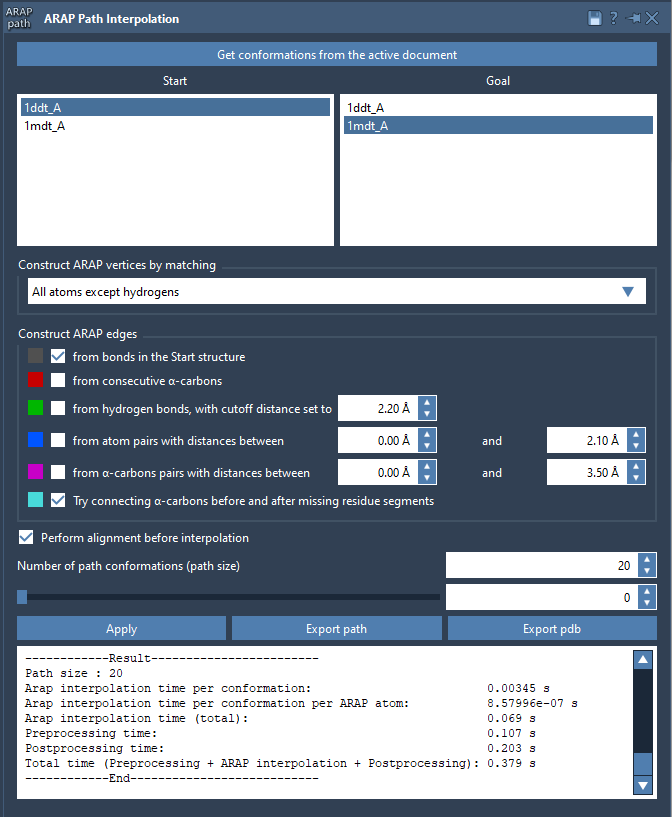
Open the interpolation app from Home > Apps > Biology > ARAP Path Interpolation. Inside the app:
- Click Get conformations from the active document
- Select
1DDT Aas start,1MDT Aas goal - Set matching atoms to All except hydrogens
- Enable alignment before interpolation
- Set number of path conformations to 20
You can define which atoms serve as ARAP vertices for controlling the interpolation geometry and also which edges (connections) guide structural coherence during interpolation.
Once the settings are defined, click Run. The generated conformations will appear as a trajectory you can scrub through using the slider.
Visualize and export
Visualize the deformation path and examine how backbone or side chain regions move during transition:
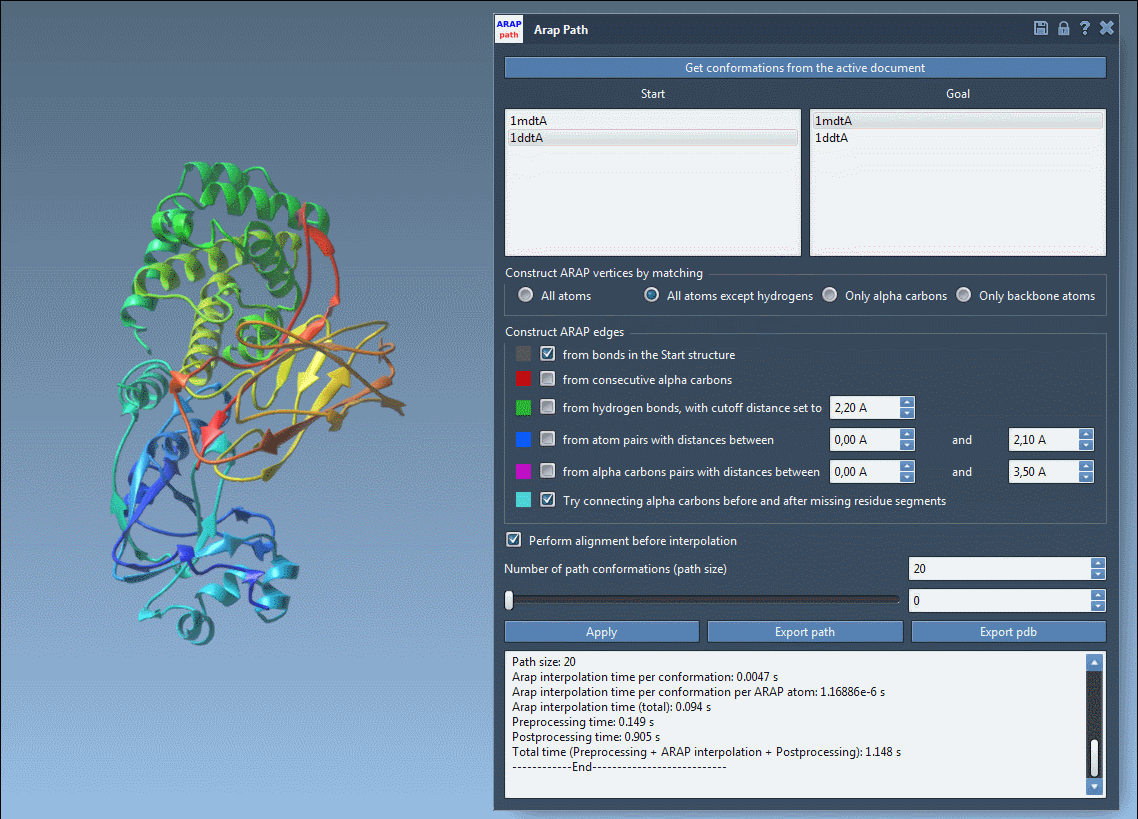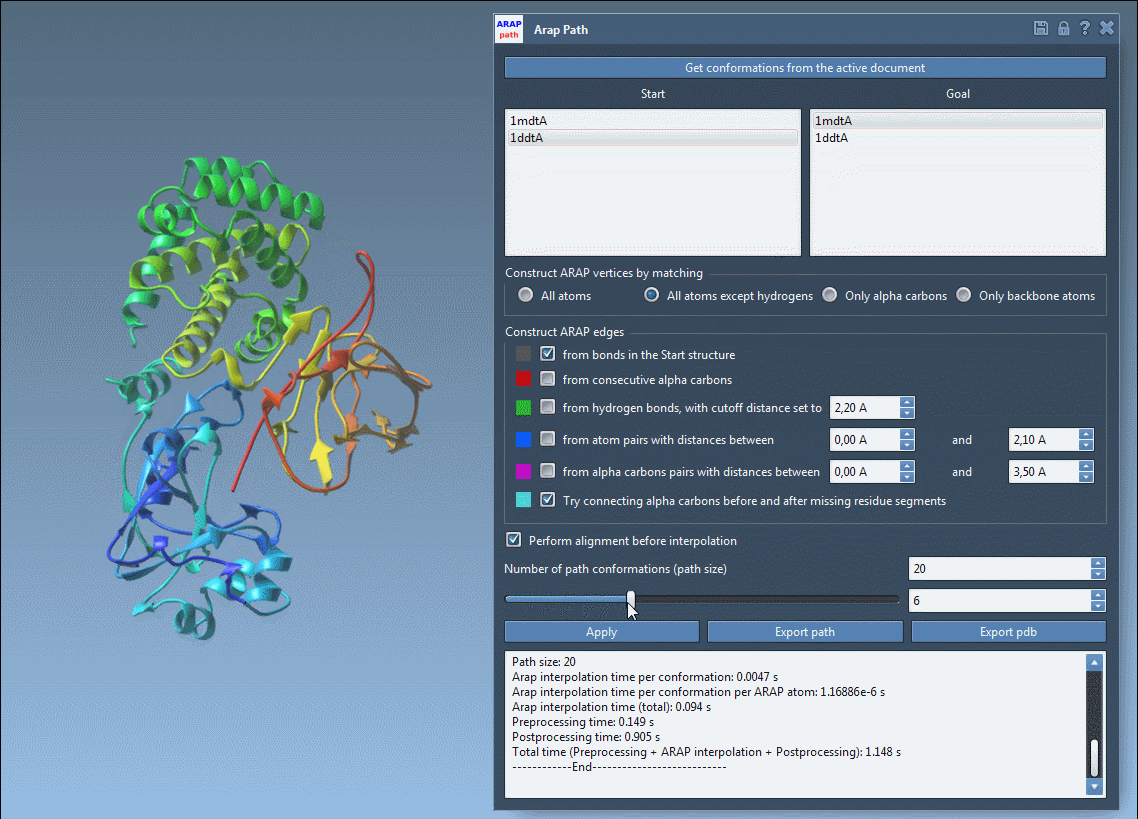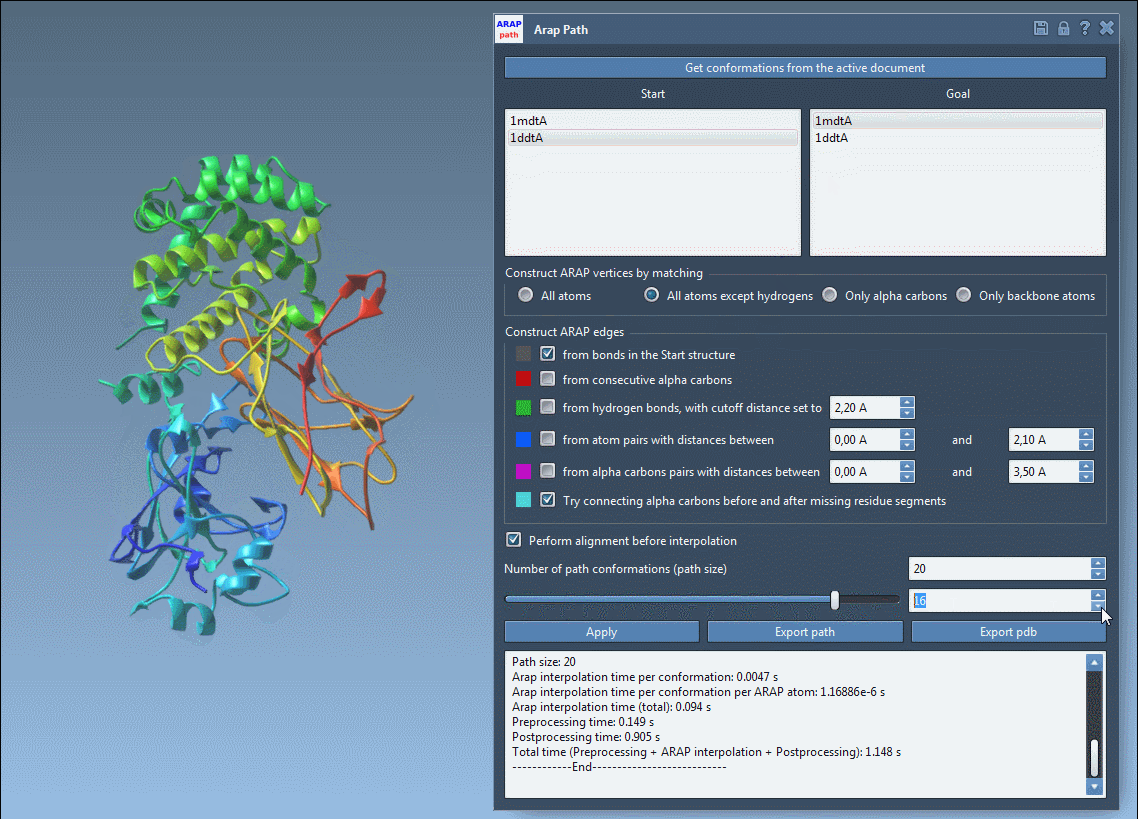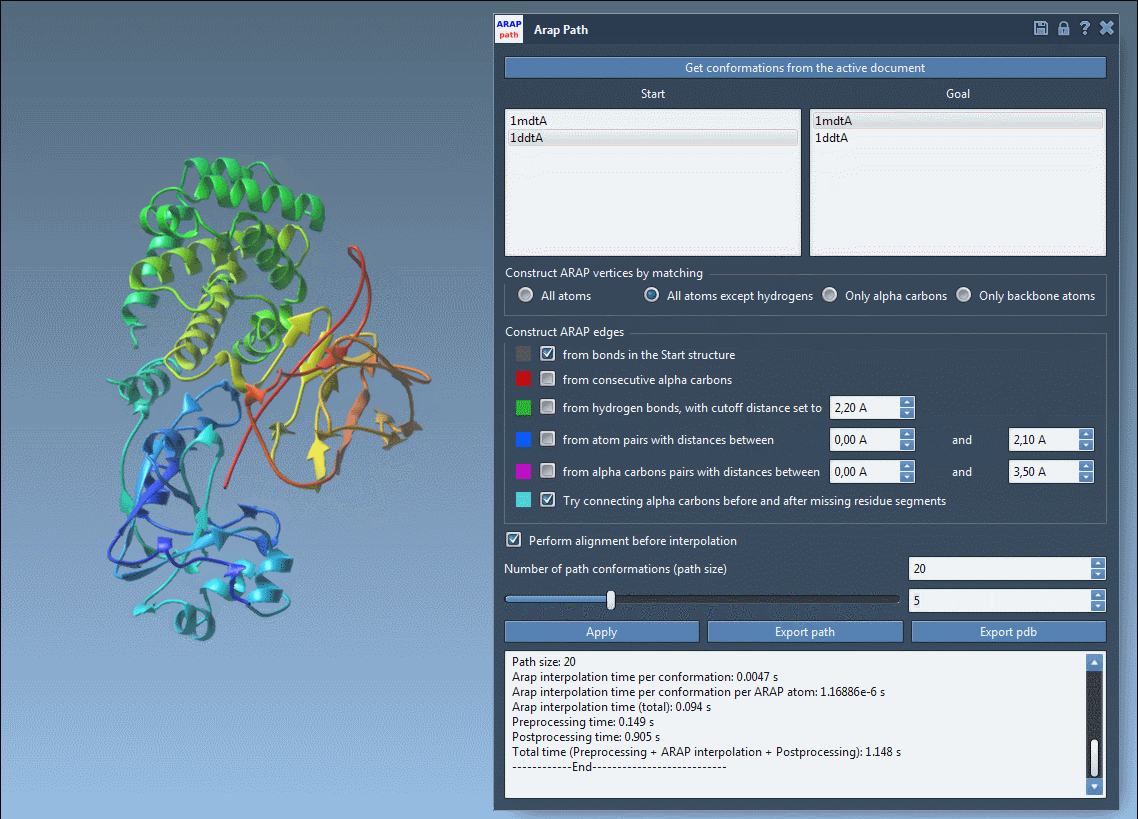
Export options let you save the trajectory as a PDB file or as individual conformations for further simulation or visualization. These structures can be reused in analysis workflows or pipelined directly into enhanced sampling applications like umbrella sampling or P-NEB path refinement.
Conclusion
If you want to analyze structural transitions without launching a molecular dynamics simulation every time, try out the ARAP Interpolator in SAMSON. It provides fast and biologically-meaningful transition paths between experimentally determined protein states—and can serve as a smart first step in many modeling workflows.
To learn more, visit the original ARAP Interpolator documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.