





Molecular modelers often face the challenge of comparing proteins to identify conserved residues, understand structural similarities, or build homology models. Accurately aligning protein sequences and structures is crucial for gaining biological insights, but finding a fast, reliable, and streamlined way to achieve this can be difficult. That’s where SAMSON’s Protein Aligner extension comes in: it provides an all-in-one workflow for aligning protein sequences, inspecting conserved residues, and superimposing structures.
In this guide, we’ll walk you through the essential steps to align protein sequences and structures using SAMSON. By the end of this article, you’ll understand how the Protein Aligner can simplify your research and analysis workflow.
Why Align Protein Sequences and Structures?
Protein alignment goes beyond just comparing sequences. It allows researchers to:
- Identify conserved residues that play a role in function or ligand binding.
- Compare structural conformations across species or protein variants.
- Assist in creating accurate homology models for downstream molecular design tasks.
SAMSON’s Protein Aligner integrates these capabilities into one interface, avoiding the need for separate tools and multiple file conversions.
Preparing Your Proteins
Before diving into sequence alignment, it’s crucial to prepare your structures. Here’s a quick checklist to ensure everything is ready:
- If your protein structures contain extra solvent or ligands, clean them using SAMSON’s Protein Preparation & Validation tool. This will help avoid alignment issues.
- Ensure you have fetched or opened the protein structures you wish to align. For instance, you could align two hemoglobins (e.g.,
1DLWand1RTX).

Fetching PDB files is intuitive within SAMSON. Simply navigate to Home > Fetch, input the desired protein IDs, and press Load.
Launching the Protein Aligner
Once your structures are prepared, you’re ready to use the Protein Aligner. Open it by selecting Home > Align from the menu:

Aligning Protein Sequences
The Protein Aligner offers two options for sequence alignment:
- Align sequences (by structure): This aligns one model to another in terms of their overall structure.
- Align sequences (by chain): This aligns individual chains, which is useful for oligomers.
Click on the corresponding Align sequences button to generate the alignment. You can toggle Highlight residues to view conserved regions based on amino acid properties, offering insights into functional or structural conservation:

Interacting With the Alignment
Once the sequences are aligned, you can interact with them to gain further insights. Hover over residues to see their names and IDs, or click residues to select them and highlight their aligned partners. Use keyboard shortcuts like Shift for selecting consecutive residues and Ctrl/Cmd to select non-consecutive residues. This level of interactivity makes it easier to analyze specific regions of interest.

Superimposing Protein Structures
SAMSON’s Protein Aligner doesn’t just stop at sequence alignment. It enables you to superimpose protein structures, which is essential when comparing structural conformations. Simply click the Align to this button on the reference model. The RMSD (Root Mean Square Deviation) between the structures will be displayed, helping you gauge their structural similarity:
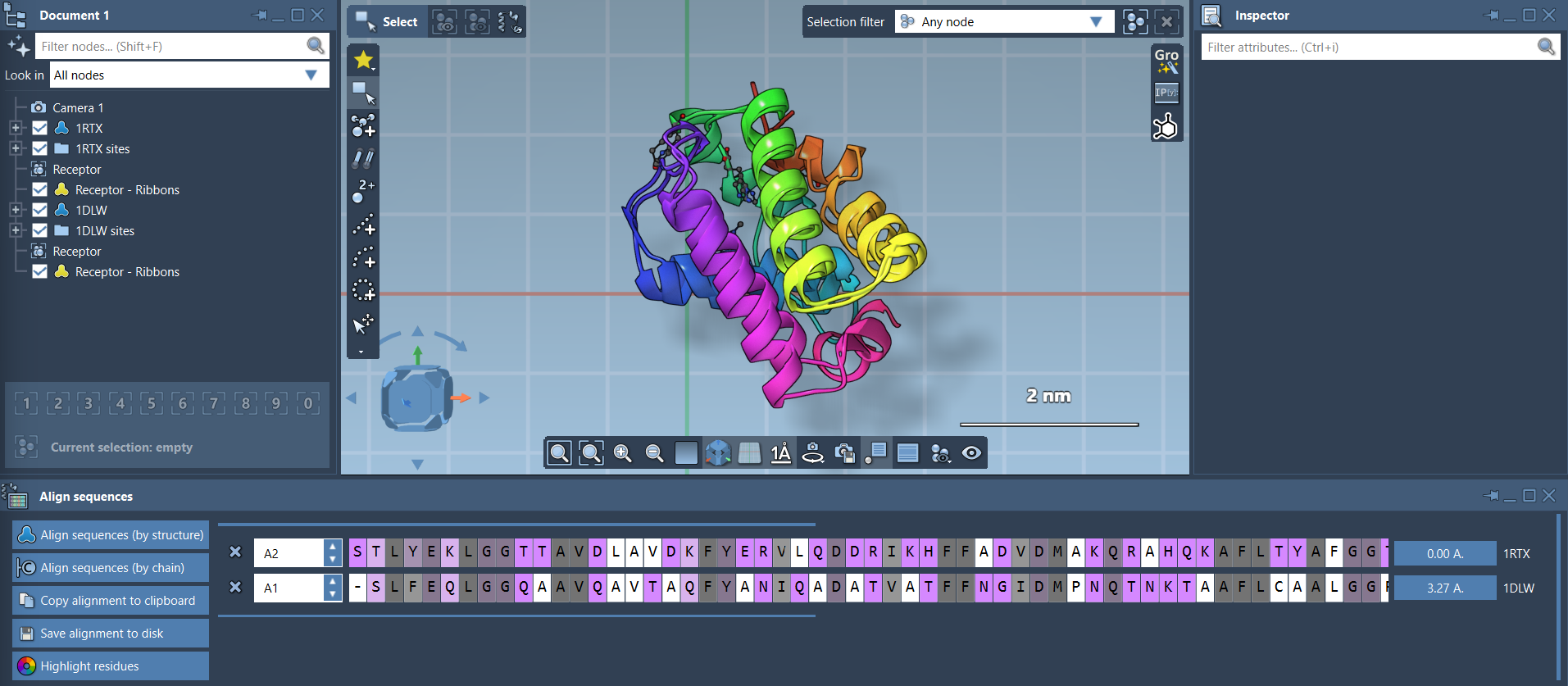
If needed, you can align specific regions of the proteins. For instance, by selecting residues of interest and clicking the alignment button, SAMSON will calculate the best superposition for those parts.
Next Steps
After aligning your proteins, you can:
- Export alignments for homology modeling.
- Map conserved residues onto functional regions or ligand-binding sites.
- Compare additional chains or sequence variants.
SAMSON makes aligning protein sequences and structures straightforward, reducing the complexity of integrating data from various tools. To explore this feature in depth, visit the official documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get started by downloading SAMSON at this link.