Author: OneAngstrom
Simplify Transition Path Optimizations with P-NEB in SAMSON
Molecular modelers often face the challenge of efficiently optimizing transition paths between two states of a system. These paths are crucial for understanding molecular mechanisms, such as ligand binding or unbinding, conformational changes, and catalytic processes. However, generating a realistic…
Understanding Light Attributes in SAMSON’s Node Specification Language
Mastering the Conceal Atoms Animation in Molecular Design
Streamlining Protein-Ligand Docking in SAMSON Using the FITTED Suite
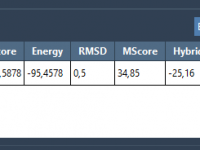
Molecular modelers frequently encounter challenges when preparing and optimizing complex systems for accurate protein-ligand docking. Time-consuming preparation steps, such as cleaning up structural files, optimizing ligand geometry, and setting up flexible docking workflows, can be a bottleneck in the design…
Streamlining Production MD Simulations: Setting Up Inputs in GROMACS Wizard
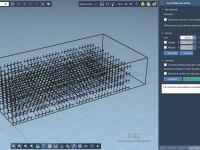
Streamline Molecular Modeling by Populating 3D Boxes in Seconds
Streamline Molecular Visualization with Visual Presets in SAMSON.
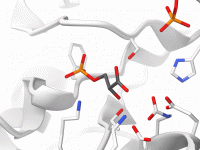
Molecular modelers face numerous challenges when visualizing complex molecular systems. Whether showcasing a protein-ligand interaction, preparing a presentation, or simply analyzing key structural elements, much time can be wasted manually applying individual visualization settings. This post offers a solution: visual…