Author: OneAngstrom

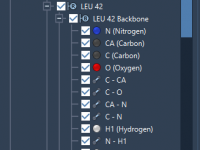
Understanding Presentation Attributes in SAMSON’s Node Specification Language
Mastering Side Chain Attributes in SAMSON’s Node Specification Language
Demystifying Path Attributes in SAMSON’s NSL
For molecular modelers, managing paths and their specific attributes in structural models can be a crucial yet time-consuming challenge. Whether you’re working on conformational analysis, interaction studies, or structural refinement, efficiently querying path-related data can be transformative. Enter SAMSON’s Node…
Streamline Molecular Modeling with Positional Analogue Scanning in SAMSON.
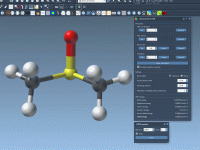
Mastering the Reveal Atoms Animation in Molecular Modeling
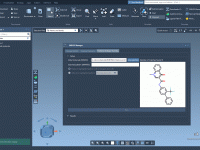
Simplifying Molecular Dynamics with Efficient Parameter Selection in GROMACS Wizard.
Creating Smooth Transitions with the Disappear Animation in SAMSON
For molecular modelers, presenting dynamic, visually engaging transitions can make molecular demonstrations more impactful and comprehensible. A common challenge arises when certain visualization elements need to gradually disappear or fade away without abrupt visual disruptions. Fortunately, SAMSON’s Disappear animation provides…