





Author: OneAngstrom
A Quick Guide to SAMSON’s Interactive Tutorials.
Enhancing Molecular Transition Paths with P-NEB in SAMSON

For molecular modelers seeking to understand the transitions between conformations in a system, optimizing transition paths is often a critical task. Whether you’re exploring ligand unbinding pathways or studying molecular dynamics, defining energy-efficient paths between conformations can be challenging. This…
Mastering the Animator in SAMSON for Powerful Molecular Presentations.
Creating clear, engaging, and visually compelling molecular presentations can often be a daunting task for molecular modelers. Whether you’re trying to illustrate complex interactions, highlight results, or make your research stand out, animations and presentations are vital tools. Fortunately, the…
Visualizing the SARS-CoV-2 Spike Protein in Motion

Streamlining Molecular Research with SAMSON’s Document Sharing Features

Master Vertical Camera Movements with the Pedestal Camera Animation
Simplify Molecular Models: Renumbering Residues and Chains for Coarse-Grained Modeling

Mastering Camera Features to Streamline Your Molecular Modeling
Master Molecular Design with SAMSON’s Interactive Tutorials
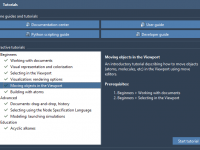
For molecular modelers, learning a new platform for molecular design can sometimes feel overwhelming, particularly with intricate tools and specialized workflows. If you’ve recently started exploring SAMSON, the platform’s Interactive Tutorials can be a game-changer, providing hands-on guidance to streamline…