Author: OneAngstrom
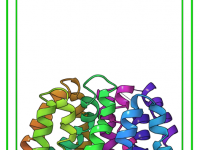
Effortless Molecular Visualizations with Visual Presets in SAMSON
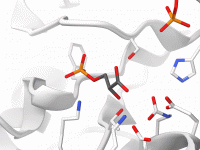
For molecular modelers, creating clear, compelling visualizations of complex molecular structures can be a daunting task. Analyzing intricate systems and presenting them effectively often requires toggling between multiple visualization tools, color schemes, and rendering styles—a tedious process that can consume…
Demystifying Camera Attributes in Molecular Modeling
Demystifying Property Model Attributes in Molecular Design
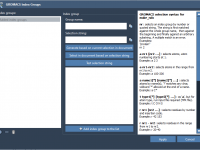
Simplify Molecular Simulations with a Custom GROMACS Setup in SAMSON
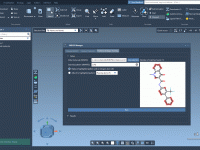
Streamlining Molecular Design with Positional Analogue Scanning in SAMSON
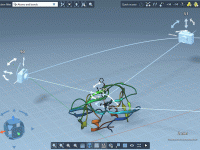
Mastering Constrained Simulations with SAMSON’s Simulate Animation
Constrained simulations are critical for molecular modelers who design nanosystems and need to explore how molecules behave within specific parameters. Creating a realistic simulation that replicates these constraints can often be a complex challenge. SAMSON’s Simulate animation provides an effective…