Author: OneAngstrom
A Better Way to Animate Atoms: Avoid Rebuilding Scenes Frame by Frame
A Simple Way to Select Atoms with Mathematical Precision

Protein folding in minutes: AlphaFold-2 predictions in SAMSON
Precisely Orient Molecular Fragments in SAMSON for Cleaner Models
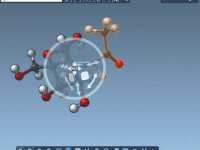
Improving Ligand Unbinding Pathways with P-NEB in SAMSON
For molecular modelers working with complex ligand-protein interactions, understanding unbinding processes often means exploring energy landscapes – and that usually includes optimizing transition paths. This is where SAMSON’s Parallel Nudged Elastic Band (P-NEB) app comes in. If you’ve generated unbinding…