Author: OneAngstrom
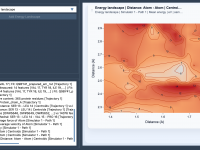
Understanding Molecular Trajectories with Path Analyzer.
Mastering Undo and Redo in Molecular Modeling
Easily Extend SAMSON with Powerful Extensions.

Understanding Conformation Attributes in SAMSON’s Node Specification Language
Personalizing the SAMSON Interface for Efficiency
Optimizing Vertical Movement with the Pedestal Camera Animation in SAMSON
Molecular modelers often find themselves in need of precise control over their camera’s perspective when presenting complex systems or navigating vertically through molecular models. If you’ve ever struggled to gracefully showcase vertical transitions, SAMSON’s Pedestal camera animation could be the…