





When working with protein conformations, it’s common to want to generate a smooth transition pathway between two structures—say, for insight into functionally relevant motions or for use in simulations like umbrella sampling. But here’s a common blocker that molecular modelers often encounter, even before the interpolation step: the structures aren’t ready to be interpolated. 🧬
If you’ve ever tried to compute a morphing path between two conformations (for example with As-Rigid-As-Possible (ARAP) interpolation in SAMSON) and ran into confusing errors like:
“Cannot proceed because the structure does not make one connected component.”
— you’re not alone. This post shows how to avoid this situation—and save time dealing with silent errors or nonworking outputs—by correctly preparing your structures for interpolation.
Missing ligands, alternate locations, residue gaps? They get in the way
Experimental PDB files often contain water molecules, ions, small ligands, and atomic positions with alternate occupancies. These seem harmless, but they typically result in disconnected molecular graphs that prevent interpolation algorithms like ARAP from working correctly.
This is because ARAP treats the structure as a network of connected atoms: it builds a graph where atoms are nodes and bonds are edges. If the system is fragmented, the algorithm can’t define this graph properly. Result: no interpolation path.
So what’s the fix?
The solution is to clean your structures beforehand using SAMSON’s Prepare function:
- Go to Home > Prepare.
- This step removes water, ligands, ions, and alternate locations.
- Use it for both your start and goal conformations.
Once cleaned, your structures become suitable for downstream workflows like conformer definition and ARAP interpolation.
Visual: Before Running Interpolation
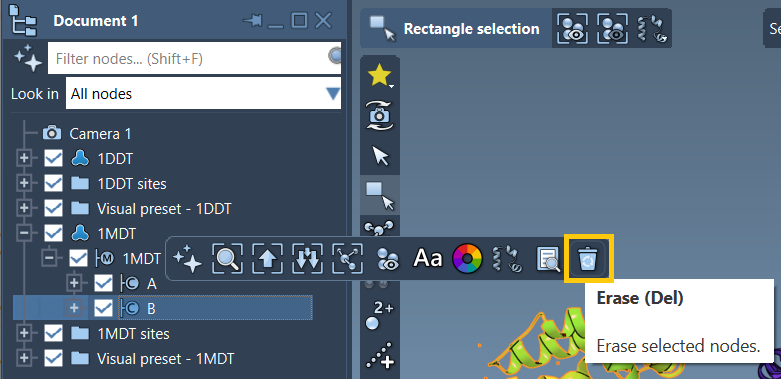
Here’s what your loaded proteins might look like before cleaning. For example, the 1MDT structure has an extra chain (B) that you’ll want to remove to ensure proper interpolation.
Quick tip: Delete unwanted chains manually
In addition to Prepare, make sure you isolate the correct chains you’re interested in interpolating. If you’re working only with chain A, delete chain B:
- Expand the structure in the Document view.
- Select and delete chain B using the Del key or the erase tool.
Common Misstep: Skipping Preparation
Not cleaning the structures before creating conformers often results in disconnected components. Unfortunately, this may not be obvious until later in the interpolation process—costing time and leading to avoidable debugging.
If you forget to run Prepare, you’ll be greeted with the aforementioned error message. So make structure preparation a habit—it only takes a click, and it saves a lot of time. 🧹
Want to go further?
Once your structures are clean, you’re ready to define conformations and use the ARAP Interpolator in SAMSON to generate fast, realistic transition pathways between protein states.
To learn more about the full workflow, including the interpolation and visualization steps, check out the official documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at samson-connect.net.