





One commonly overlooked yet crucial step in molecular docking studies is ligand preparation. 🧪 Docking algorithms like AutoDock Vina assume that ligands are clean, chemically sensible, and ready to interact with the target. Unfortunately, that’s not always the case — especially when ligands come from diverse libraries.
Improperly prepared ligands can lead to bad scores, unrealistic poses, or even failed docking runs. This is where the AutoDock Vina Extended extension in SAMSON comes in handy — not only does it let you dock ligands, but it also guides you through proper setup. Let’s walk through some key features that can help you prepare ligands effectively and avoid common pitfalls.
Detect and Fix Rotatable Bonds
After selecting your ligand in the Document view and assigning it in the “Set ligand” section, you’ll immediately see green cylinders representing rotatable bonds:
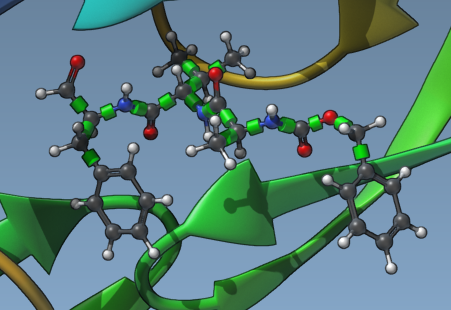
Clicking on these controllers toggles their state: green (rotatable) or red (locked). It’s a highly intuitive visual system — and enables you to fine-tune how flexible your ligand should be during docking.
Lock Unwanted Bond Rotations
To ensure specific types of bonds remain rigid, SAMSON lets you configure Locked bonds settings first, and then enforce them globally using the Lock specific ligand bonds option:
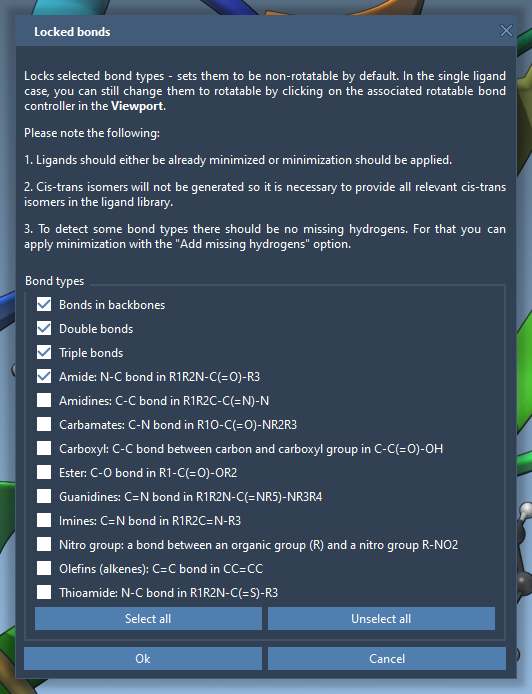
This helps preserve geometry for functional groups that shouldn’t rotate—like aromatic rings or double bonds—and ensures the docking stays chemically meaningful.
Don’t Dock Flat Ligands: Minimize First
Many ligand libraries (especially public ones like ZINC) contain 2D or poorly optimized structures. These structures may misbehave during docking unless pre-optimized.
Fortunately, SAMSON includes a ligand minimization feature directly in the AutoDock Vina Extended interface:

With just a checkbox, you can ensure each ligand undergoes energy minimization prior to docking. You can even specify presets for convergence thresholds and whether to add hydrogens if they’re missing.
Hydrogens: Add When Missing, Leave When Validated
AutoDock Vina requires polar hydrogens for hydrogen bond inference. If your ligands don’t already include them, checking the Add hydrogens box during minimization saves you the hassle of doing it manually. However, note that if your hydrogens reflect a validated protonation state for a specific pH, you may prefer to leave them unchanged.
Lock Cis/Trans Isomers Into Place
It’s important to know that cis/trans isomers won’t be automatically generated during docking. So, if isomer specificity matters to your study (e.g., for pharmacophore reasons), make sure your ligand library has all relevant isomeric forms ahead of time.
A small side note: Locked bonds affect docking — but not minimization. Even locked bonds will rotate during ligand minimization if it leads to lower energy.
Wrapping Up
Taking a few extra minutes to carefully set up your ligands pays off in more reliable and interpretable docking outcomes. SAMSON’s visual bond controllers, minimization options, and bond locking settings provide powerful tools to get this right — without the steep learning curve 🎯
To explore the full tutorial, visit the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.