Exploring complex molecular transformations often means working with trajectories that capture conformational changes or dynamic processes. One of the common challenges molecular modelers face is presenting those trajectories in an intuitive and synchronized way — especially when paths don’t align…
Anyone working with molecular models knows the slow drip of time lost to repetitive cleanup tasks: removing waters, deleting unwanted ligands, fixing missing atoms, and ensuring the structure won’t crash a simulation. It’s manageable when working with one or two…
When working on complex molecular models, it’s common to ask very specific questions about a particular system: How many backbone groups have a partial charge above a certain threshold? Which ones are invisible or lack material? Are there backbones with…
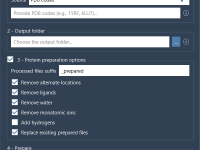
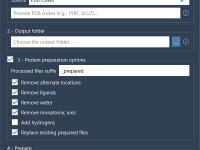
If you’ve ever had to prepare a large number of protein structures for simulations or docking runs, you know how time-consuming it can be to check alternate locations, add missing atoms, strip solvents, and standardize formats—file by file. Whether you’re…
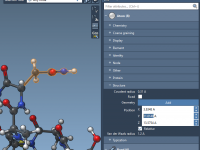
One of the recurring challenges faced by molecular modelers is fine-tuning the structural details of their models without introducing inconsistencies. Whether you’re adjusting atomic positions, types, or making minor corrections in a structure, you often want surgical precision over your…
In molecular modeling, presenting a complex mechanism often requires temporarily hiding parts of a molecular system to direct attention. For example, a protein’s active site might be buried deep within a folded structure. To highlight its dynamics, it helps to…
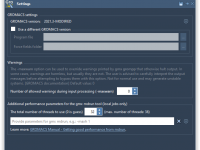
Running molecular dynamics simulations can be demanding on your hardware. It’s common to see users overwhelmed by lagging systems or jobs that fail due to poor performance settings. If you’re using GROMACS Wizard in SAMSON, there’s a simple way to…
Imagine you’re preparing to simulate a protein in a new solvent environment. One of the first steps is deciding which residues might interact more strongly with the environment—perhaps charged residues for pH-sensitive behavior, or polar residues for solvent-accessible surface analysis.…
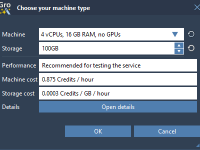
Molecular dynamics (MD) simulations can be time-consuming, especially when you’re working with larger systems or limited local computational power. For many molecular modelers, long simulations mean one thing: your workstation becomes unusable for hours, or even days. Luckily, SAMSON GROMACS…
When dealing with complex molecular systems, it’s often necessary to focus on specific parts of a model to streamline analysis or editing. For example, you might want to target all molecular paths with more than 100 atoms for further inspection,…