





Category: Uncategorized
Finding Molecular Backbones by Property in SAMSON
Preparing Dozens of Protein Structures? Try This Workflow-Saving Tool
Changing Atom Attributes Without Breaking Your Model
Let Keyframes Do the Hiding: A Simple Way to Control Molecular Visibility in SAMSON
Using GROMACS Performance Parameters Without Overloading Your System
Quickly Select Charged or Polar Residues Using NSL in SAMSON
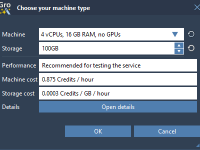
Making the Most of Idle Time: Run GROMACS Simulations in the Cloud
How to Quickly Select Molecular Paths with a Specific Number of Atoms in SAMSON
Avoid Chain Conflicts When Generating CG Models with Martinize2
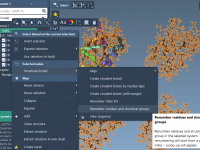
When working with coarse-grained (CG) molecular models, it’s common to simulate multiple copies—or replicas—of the same protein in a single system. This setup is especially useful for studies involving protein aggregation, diffusion, or encapsulation. However, many researchers new to coarse-graining…
Finding Molecular Backbones by Property in SAMSON
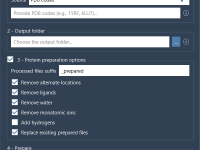
Preparing Dozens of Protein Structures? Try This Workflow-Saving Tool
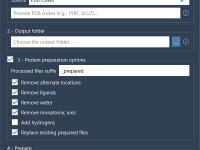
Changing Atom Attributes Without Breaking Your Model
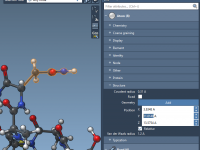
Let Keyframes Do the Hiding: A Simple Way to Control Molecular Visibility in SAMSON
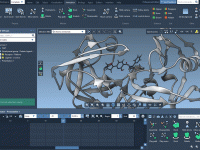
Using GROMACS Performance Parameters Without Overloading Your System
Quickly Select Charged or Polar Residues Using NSL in SAMSON
Making the Most of Idle Time: Run GROMACS Simulations in the Cloud
How to Quickly Select Molecular Paths with a Specific Number of Atoms in SAMSON
Avoid Chain Conflicts When Generating CG Models with Martinize2
When working with coarse-grained (CG) molecular models, it’s common to simulate multiple copies—or replicas—of the same protein in a single system. This setup is especially useful for studies involving protein aggregation, diffusion, or encapsulation. However, many researchers new to coarse-graining…